Болезнь Краббе (глобоидно-клеточная лейкодистрофия)
Что провоцирует / Причины Болезни Краббе (глобоидно-клеточной лейкодистрофии):
Патогенез (что происходит?) во время Болезни Краббе (глобоидно-клеточной лейкодистрофии):
В головном мозге, печени, селезенке, почках, лейкоцитах, фибробластах накапливаются галактоцереброзид и его производное психозин. Количество последнего повышается в 10-100 раз, что оказывает токсическое воздействие на олигодендроглиальные клетки, формирующие миелиновую оболочку. В зонах демиелинизации вокруг мелких кровеносных сосудов белое вещество содержит большое количество глобоидных гистиоцитов (макрофагов). Уменьшение олигодендроглиальных клеток сопровождается глиозом. Периферические нервы подвергаются аксональной дегенерации с накоплением пенистых гистиоцитов.
Симптомы Болезни Краббе (глобоидно-клеточной лейкодистрофии):
Как правило, первые клинические симптомы появляются на четвертом месяце жизни. Отмечаются повышенная возбудимость и мышечная гипертония. Верхние и нижние конечности у больных детей находятся в разогнутом состоянии, кулаки сжаты. Вскоре становится заметной задержка психомоторного развития. Позднее развиваются миоклонические судороги, генерализованная двигательная реакция на слуховые раздражители, спастический тетрапарез, повышение глубоких сухожильных рефлексов, при исследовании глазного дна выявляют атрофию зрительных нервов. Проявления периферической нейропатии встречаются лишь в отдельных случаях. В последующем развиваются характерные глубокие умственные расстройства, вялый тетрапарез, снижаются сухожильные рефлексы, мышечная гипертония сменяется гипотонией. Больные умирают в возрасте от 7 месяцев до 3 лет.
Диагностика Болезни Краббе (глобоидно-клеточной лейкодистрофии):
Диагноз основан на клинической картине, повышении содержания белка в цереброспинальной жидкости, выявлении снижения активности галактозилцерамид-b-галактозидазы. Дифференциальный диагноз проводят с другими формами лейкодистрофии на основании биохимического анализа.
Лечение Болезни Краббе (глобоидно-клеточной лейкодистрофии):
Специфическое лечение не разработано. Возможна пренатальная диагностика болезни Краббе путем исследования в культивируемых амниотических клетках активности галактозилцерамид-b-галактозидазы.
К каким докторам следует обращаться если у Вас Болезнь Краббе (глобоидно-клеточная лейкодистрофия):
Болезнь Краббе: причины, симптомы и методы лечения

Лекарства от болезни Краббе не существует, терапия проводится в основном для поддержания состояния пациента. Однако исследования показали эффективность трансплантации стволовых клеток младенцам, которым была проведена операция до появления первых симптомов.
Симптомы болезни Краббе
В большинстве случаев признаки и симптомы болезни Краббе проявляются в течение первых 2-5 месяцев жизни.
Младенцы
Общие признаки и симптомы на ранних стадиях заболевания включают в себя:
По мере прогрессирования заболевания
Признаки и симптомы становятся все более выраженными со временем. Они могут включать в себя:
Дети старшего возраста и взрослые
Когда болезнь Краббе развивается позже в детстве или во взрослом возрасте, признаки и симптомы могут сильно варьироваться. Они могут включать в себя:
Как правило, чем моложе возраст, в котором возникает глобоидно-клеточная лейкодистрофия, тем быстрее болезнь прогрессирует и тем более вероятен летальный исход.
У некоторых людей, у кого заболевание было диагностировано в подростковом или взрослом возрасте, в основном наблюдается только мышечная слабость, и нет никакого ухудшения мыслительных навыков.
Когда следует обратиться к врачу?
Ранние признаки болезни Краббе в младенчестве могут указывать на любое заболевание или проблемы со здоровьем. Именно поэтому важно получить быстрый и точный диагноз.
Симптомы, чаще всего связанные с более старшими детьми и взрослыми, также не являются специфичными для болезни Краббе и требуют своевременной диагностики.
Причины болезни Краббе
В случае с глобоидно-клеточной лейкодистрофией две мутировавшие копии определенного гена приводят к незначительному или полному отсутствию продукции фермента, называемого галактоцереброзидазой (GALC). Он отвечает за расщепление определенных веществ в центре рециркуляции клетки (лизосоме).
При болезни Краббе недостаток ферментов GALC приводит к накоплению определенных типов жиров, называемых галактолипидами. Они обычно существуют в клетках, производящих и поддерживающих защитное покрытие нервных клеток (миелин). Однако обилие галактолипидов оказывает токсическое действие, приводящее к саморазрушению миелинообразующих клеток.
Потеря миелина (демиелинизация) мешает нервным клеткам посылать и получать сообщения.
Фактор риска
Мутация гена, связанная с болезнью Краббе, вызывает заболевание только в случае наследования двух мутировавших копий гена. В этом случае говорят про аутосомно-рецессивное расстройство.
Если у каждого родителя есть одна мутировавшая копия гена, то риск для ребенка будет следующим:
Именно поэтому, если в паре оба родителя являются носителями мутации гена GALC, может потребоваться пренатальный генетический тест для понимания риска рождения ребенка с болезнью Краббе.
Осложнения, вызванные болезнью Краббе
У детей с прогрессирующей болезнью Краббе может развиться целый ряд осложнений, включая инфекции и респираторные заболевания. На поздних стадиях заболевания ребенок становится недееспособным, прикованным к постели и в конце концов впадает в вегетативное состояние.
Большинство детей, у которых развивается болезнь Краббе в младенчестве, умирают до 2 лет, чаще всего от дыхательной недостаточности или осложнений неподвижности и заметно сниженного мышечного тонуса.
Дети, у которых болезнь развивается позже, могут иметь несколько большую продолжительность жизни, обычно между двумя и семью годами после постановки диагноза.
Диагностика болезни Краббе
Диагностика болезни Краббе основывается на серии тестов, которые могут включать в себя следующее.
Для оценки уровня активности фермента GALC используется образец крови. Очень низкий или вообще отсутствующий показатель может указывать на болезнь Краббе. Однако не всегда низкая активность GALC означает, что состояние будет прогрессировать.
Для обнаружения потери миелина в пораженных областях мозга врач может назначить один или несколько тестов визуализации, среди которых МРТ и КТ.
Исследование нервной проводимости оценивает скорость, с которой нервы передают сигнал — по существу, как быстро они могут отправить сообщение. Специальное устройство измеряет время, необходимое электрическому импульсу для перемещения из одной точки тела в другую. При нарушении функции миелина нервная проводимость замедляется.
Для подтверждения диагноза используется генетический тест, который также поможет определить ожидаемое течение заболевания.
Начальный скрининговый тест измеряет активность фермента GALC. Если обнаруживается низкая активность фермента, проводятся последующие GALC-тесты и генетические тесты.
Исследования, проведенные на сегодняшний день, показывают, что выявление маркеров болезни Краббе до появления симптомов может создать уникальное окно для лечения.
Процедура лечения, называемая трансплантацией стволовых клеток, может улучшить течение глобоидно-клеточной лейкодистрофии при проведении в первые недели жизни.
Лечение болезни Краббе
Для младенцев, у которых уже развились симптомы болезни Краббе, в настоящее время не существует лечения, которое могло бы изменить течение состояния. Терапия, таким образом, фокусируется на управлении симптомами и оказании поддерживающей помощи.
Мероприятия могут включать в себя:
Мероприятия для детей старшего возраста или взрослых с менее тяжелыми формами заболевания могут включать в себя:
Современные данные свидетельствуют о том, что трансплантация стволовых клеток наиболее эффективна, когда она проведена до достижения ребенком двухнедельного возраста. Исследования показали, что у малышей с предсимптомной стадией заболевания, прошедших трансплантацию стволовых клеток, возможно более медленное прогрессирование заболевания и улучшение качества и продолжительности жизни.
Однако у младенцев, перенесших трансплантацию стволовых клеток до появления симптомов заболевания, все еще развиваются значительные трудности с речью, ходьбой и другими двигательными навыками в детстве.
Болезнь краббе это что простыми
Болезнь Краббе — редкое нейродегенеративное заболевание с аутосомно-рецессивным типом наследования, выраженным распадом миелина и глобоидными тельцами в белом веществе мозга. Ген, определяющий развитие болезни Краббе (GALC), локализован на хромосоме 14q-24.3-q32.1. В основе заболевания лежит выраженный дефицит лизосомного фермента галактоцереброзид-b-галактозидазы, который отщепляет часть молекулы галактозы от церамидной порции галактоцереброзида. Болезнь Краббе — заболевание с деструкцией миелина, а не с формированием аномального миелина.
В норме миелинизация начинается в III триместре беременности, что соответствует быстрому увеличению активности галактоцереброзид-b-галактозидазы в головном мозге. У пациентов с болезнью Краббе метаболизм галактоцереброзида в процессе нормального обмена миелина невозможен в связи с дефицитом галактоцереброзид-b-галактозидазы. При введении галактоцереброзида в мозг экспериментальных животных наблюдается глобоидноклеточная реакция. Установлено, что сходный феномен имеет место и у людей: неметаболизирующийся галактоцереброзид стимулирует образование глобоидных клеток, что отражает деструкцию клеток олигодендроглии.
Поскольку олигодендроглиальные клетки отвечают за выработку миелина, гибель этих клеток приводит к распаду миелина, в результате которого образуется дополнительное количество галактоцереброзида; таким образом, формируется порочный круг деструкции миелина.
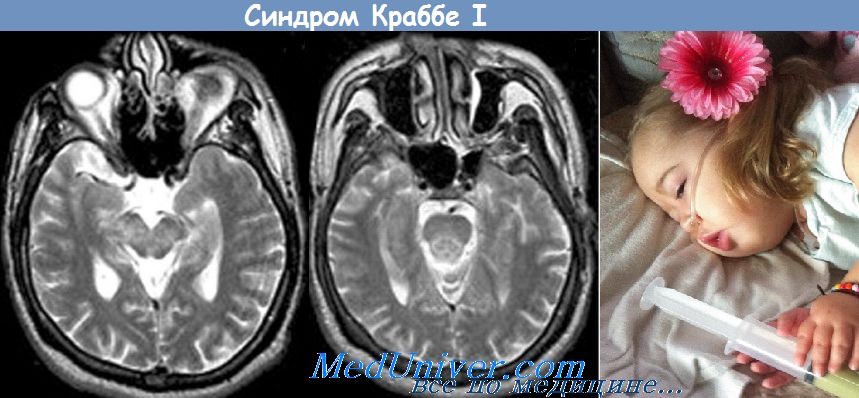
Симптомы болезни Краббе проявляются в первые несколько месяцев жизни и включают чрезмерную раздражительность и частый плач, необъяснимые эпизоды гиперпирексии, нарушение питания, рвоту и задержку прибавки массы тела. На первой стадии заболевания у детей часто диагностируется колика или аллергия на молоко, проводится соответствующее лечение с частым изменением формулы молочных смесей.
Генерализованные судороги могут возникать на ранней стадии. Изменение мышечного тонуса с развитием ригидности и опистотонуса, отсутствие зрительного внимания вследствие атрофии зрительных нервов появляются по мере прогрессирования заболевания. В поздней стадии основу клинической картины составляют такие симптомы, как слепота, глухота, отсутствие глубоких сухожильных рефлексов и децеребрационная ригидность. На КТ головного мозга без контрастирования возможно симметричное повышение плотности в области хвостатых ядер и таламуса. Большинство пациентов умирают к 2-летнему возрасту.
Описана болезнь Краббе с поздним дебютом — в школьном или подростковом возрасте. У пациентов с этим заболеванием выявляется атрофия зрительных нервов и корковая слепота, нередко в этих случаях ошибочно диагностируется АЛД. Медленно прогрессируют нарушение походки, спастичность и атаксия. Как и при классической болезни Краббе, в белом веществе большое количество глобоидных клеток, в лейкоцитах определяется дефицит галактоцереброзид-и-галактозидазы. В СМЖ повышено содержание белка, значительно снижена скорость проведения возбуждения по нервам вследствие сегментарной демиелинизации периферических нервов.
Амплитуда ЗВП постепенно уменьшается, и на поздних стадиях заболевания зрительный ответ отсутствует. При исследовании слуховых ВП ствола характерны только волны I и II. На КТ и МРТ выраженное уменьшение количества белого вещества, особенно в мозжечке и полуовальном центре (centrum semiovale), в сочетании с уменьшением количества (обеднением) субкортикальных U-волокон. Разработан метод пренатальной диагностики, основанный на определении активности галактоцереброзид-b-галактозидазы в ворсинах хориона или в культуре амниотических клеток.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Болезнь краббе это что простыми
Глобоидно-клеточная лейкодистрофия (GLD, болезнь Краббе) является аутосомно-рецессивным заболеванием, вызванным дефицитом галактоцереброзид-бета-галактозидазы (галактоцереброзидазы) (Suzuki et al., 1971). Ген находится на хромосоме 14q24—32.1. Галактоцереброзиды являются наиболее специфичными липидами миелина головного мозга; их концентрация отражает созревание. Выделяют три подтипа GLD: наиболее распространенную младенческую форму и редкие формы (ювенильную и взрослую).
Нейропатология болезни Краббе характеризуется недостаточностью олигодендроцитов, отсутствием миелина и пролиферацией глиальных клеток в пораженных участках в сочетании с мононуклеарными эпителиоидными клетками и скоплениями крупных многоядерных глобоидных клеток. Поражение периферической нервной системы встречается часто, но может быть выражено не так сильно, как при MLD. Дефицит галактоцереброзид-бета-галактозидазы приводит к накоплению цереброзидов и психозина (галактозил сфингозина). Многие симптомы, отмечаемые при GLD, связаны с повышением уровня психозина, которые оказывает токсическое действие (Suzuki, 2003).
Наиболее распространенная младенческая форма манифестирует обычно в виде гипотонии, развивающейся до шестимесячного возраста. Кроме того, характерными симптомами являются чрезвычайная возбудимость, особенно заметны вздрагивание при громких звуках и прогрессирующая ригидность. При осмотре выявляется повышение мышечного тонуса и пирамидные знаки, а также опистотонусные судороги с ретракцией головы, часто вызываемые внешним воздействием.
Во всех случаях инфантильной формы заболевания отмечается высокий уровень белка (более 70 мг/дл) в спинномозговой жидкости, а скорость проводимости нервов снижена. При формах с поздним началом уровень белка в спинномозговой жидкости может не отличаться от нормы. На МРТ в области белого вещества мозжечка выявляется снижение плотности в режиме Т1 и повышение плотности в режиме Т2. При МР-спектроскопии в пораженном белом веществе выявляется заметное повышение уровня инозит и холин-содержащих веществ, что отражает демиелинизацию и глиальную пролиферацию, сопровождающуюся снижением N-аспартил ацетата, что является признаком утраты нейронов (Brockmann et al., 2003а).
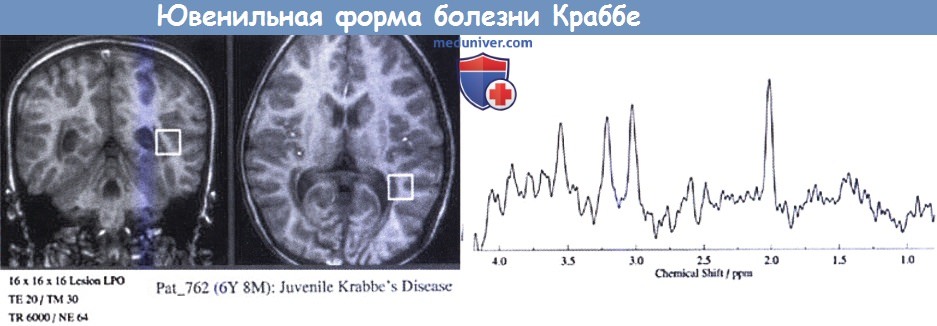 Ювенильная форма болезни Краббе.
Ювенильная форма болезни Краббе.
На МРТ (вверху) видны только небольшие аномалии сигнала белого вещества.
Заметен низкий сигнал в области таламуса.
На МР-спектроскопии (внизу) видны высокие пики холина и миоинозитола и низкие лики N-ацетиласпартовой кислоты.
Диагностика GLD основана на характерных клинических симптомах, повышении уровня белка в спинномозговой жидкости и изменениях на МРТ. При позднем начале заболевания ключевые симптомы менее характерны и включают трудности при ходьбе, снижение зрения и трудности в обучении. При любой лейкодистрофии в сочетании с легкой периферической нейропатией следует подозревать GLD. Диагноз подтверждается путем оценки уровня галактоцероброзид-бета-галактозидазы в лейкоцитах и фибробластах.
Молекулярный анализ может выявить одну из более 60 мутаций, описанных при болезни Краббе (Kleiyer et al., 1997; Wenger et al., 1997).
Лечение имеет симптоматический характер. Бензодиазепины в высоких дозах могут быть эффективны для устранения чрезвычайной возбудимости и опистотонуса у некоторых пациентов. Предпринимались попытки трансплантации стволовых гемопоэтических клеток, но полученные результаты неоднозначны (Krivit et al., 1998). В последнее время восстановление нормального уровня галактоцереброзида в крови было достигнуто у 11 новорожденных с бессимптомным течением заболевания и 14 младенцев с симптомами болезни Краббе; результаты были достигнуты благодаря трансплантации пуповинной крови неродственных доноров (Escolar et al, 2005).
У младенцев, которым трансплантация была проведена до появления симптомов, в течение всего периода наблюдения (до трех лет) отмечалось прогрессирование центральной миелинизации, развитие и приобретение навыков в соответствии с возрастом. Трансплантация, проведенная после появления симптомов, не приводила к стойкому улучшению.
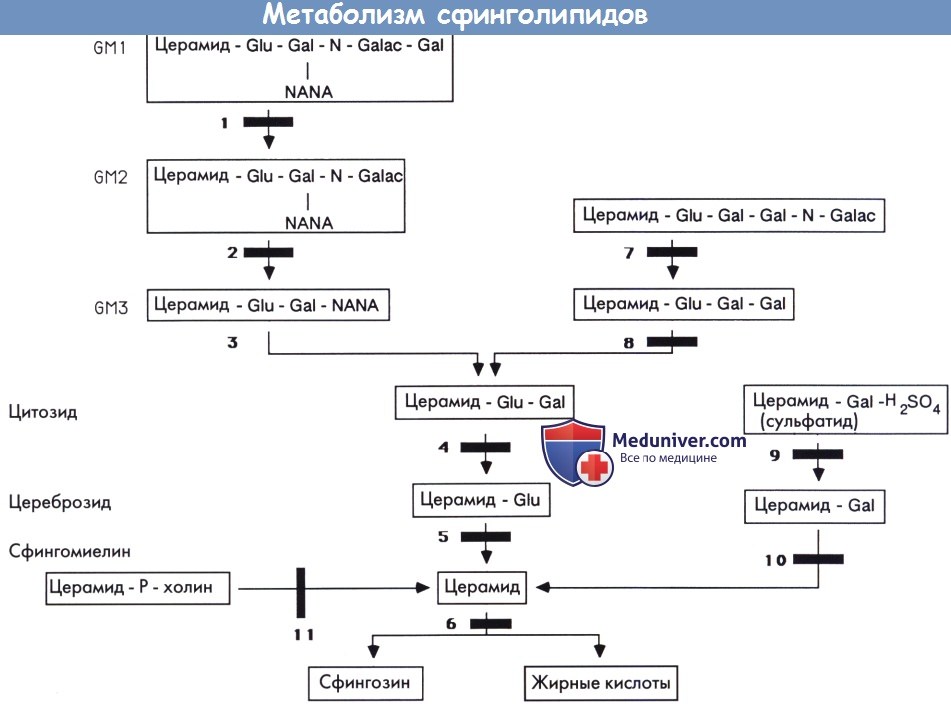 Метаболизм сфинголипидов.
Метаболизм сфинголипидов.
Gal—галактоза. Glu—глюкоза. N-Galac—N-ацетилгалактозамин. NANA—нейраминовая кислота (N-ацетилнейраминовая кислота).
1 — β-галактозидаза. 2 — β-гексозаминидаза А (болезнь Тея-Сакса) и другие ганглиозидозы типа II GМ2.
3 — GM3 сиалидаза. 4 — лактозилцекамидаза (лактозилцерамидоз). 5 — β-глюкозидаза (глюкоцереброзидаза) (болезнь Гоше).
6 — церамидаза (болезнь Фарбера). 7 — β-гексозаминидаза В (болезнь Сандгоффа и другие О варианты GM2 ганглиозидозов).
8 — церамид-тригексозидаза/α-галактозидаза (болезнь Фабри). 9 — цереброзидсульфатаза/арилсульфатаза* (метахроматическая лейкодистрофия).
10 — β-галактозидаза/β-галактоцереброзидаза (болезнь Краббе). 11 — сфингомиелиназа (болезнь Ниманна-Пика А и В).
*Активность, оцененная на искусственных субстратах, не всегда соответствовала активности цереброзиодсульфатазы.
Редактор: Искандер Милевски. Дата публикации: 16.12.2018
Публикации в СМИ
Болезнь Краббе
Болезнь Краббе — наследуемая энцефалопатия детского возраста с быстро прогрессирующей церебральной дегенерацией, демиелинизацией, проявляющаяся повышением мышечного тонуса, приступами гиперпирексии и нарушениями интеллекта. Обычно дебютирует в возрасте 3–6 мес.
Клиническая картина • Ранняя инфантильная форма (начало в 3–6 мес) •• I стадия: повышенная возбудимость ребёнка, двигательная реакция (тонические судороги) на звук, свет и т.д.; повышение мышечного тонуса, замедление развития, приступы гиперпирексии •• II стадия: развитие опистотонуса с тоническим разгибанием рук и ног, утрата всех навыков, миоклонии, судорожные припадки, гипо- или арефлексия, приступы гиперпирексии •• III стадия: децеребрация, бульбарные расстройства, судороги • Поздняя инфантильная форма: раннее развитие амавроза, постепенное нарушение интеллекта и движений.
Лечение симптоматическое.
Синонимы • Лейкодистрофия глобоидноклеточная • Краббе–Бенеке болезнь • Краббе глобоидноклеточная лейкодистрофия • Краббе диффузный инфантильный склероз
МКБ-10 • E75.2 Другие сфинголипидозы
Код вставки на сайт
Болезнь Краббе
Болезнь Краббе — наследуемая энцефалопатия детского возраста с быстро прогрессирующей церебральной дегенерацией, демиелинизацией, проявляющаяся повышением мышечного тонуса, приступами гиперпирексии и нарушениями интеллекта. Обычно дебютирует в возрасте 3–6 мес.
Клиническая картина • Ранняя инфантильная форма (начало в 3–6 мес) •• I стадия: повышенная возбудимость ребёнка, двигательная реакция (тонические судороги) на звук, свет и т.д.; повышение мышечного тонуса, замедление развития, приступы гиперпирексии •• II стадия: развитие опистотонуса с тоническим разгибанием рук и ног, утрата всех навыков, миоклонии, судорожные припадки, гипо- или арефлексия, приступы гиперпирексии •• III стадия: децеребрация, бульбарные расстройства, судороги • Поздняя инфантильная форма: раннее развитие амавроза, постепенное нарушение интеллекта и движений.
Лечение симптоматическое.
Синонимы • Лейкодистрофия глобоидноклеточная • Краббе–Бенеке болезнь • Краббе глобоидноклеточная лейкодистрофия • Краббе диффузный инфантильный склероз
МКБ-10 • E75.2 Другие сфинголипидозы