Расшифровка анализа на кариотипирование
Нормальный кариотип человека содержит 46 хромосом: 22 пары аутосом и 1 пару половых хромосом – ХХ у женщин и ХY у мужчин. Нарушение этой генетической структуры является причиной наследственных болезней, бесплодия и самопроизвольного прерывания беременности.
Плохой результат кариотипирования выглядит как:
Что значит изменение количества хромосом в кариотипе
Аномалии кариотипа приводят к порокам развития плода, поэтому такие беременности в 50-60% случаев заканчиваются выкидышем в 1 триместре. Избыток генетического материала – причина трисомий 13 (47ХХ или ХY, 13+), 18 (47ХХ или XY, 18+) и синдрома Дауна (47ХХ или ХY, 21+).
Потеря участка короткого плеча пятой хромосомы (46ХХ или ХY,5р-) вызывает синдром кошачьего крика, такой же дефект длинного плеча пятнадцатой хромосомы (46ХХ или ХY, del15q11-q13) – болезнь Прадера Вилли, признаками которой являются низкие тонус мышц и плотность костей, задержка психического и речевого развития, гипогонадизм.
Нарушения кариотипа, связанные с гоносомами, имеют менее выраженные патологические проявления: их носители могут отличаться только высоким ростом и чуть сниженным интеллектом.
Таблица расшифровки аномалий половых хромосом
| Кариотип | Заболевания |
|---|---|
| 45ХО | Синдром Шерешевского-Тернера, встречается только у женщин (1:2000 случаев). Для синдрома Тернера характерны бесплодие, рост на 20-30 см ниже среднего, короткая шея, низкое положение ушных раковин. |
| 47ХХХ | Синдром трисомии-Х, чаще всего определяется случайно, при профилактическом обследовании. Это здоровые внешне женщины с генетической склонностью к невынашиванию беременности и хромосомным аномалиям у потомков. |
| 47ХYY | Синдром полисомии-Y встречается у мужчин высокого роста (1:1000 случаев), способных зачать ребенка. Для носителей полисомии-Y характерны сниженный интеллект и склонность к агрессии. |
| 47ХХY | Синдром Клайнфельтера – мужская патология (1:500 случаев), может проявляться в виде азооспермии, отсутствия сперматозоидов в эякуляте и бесплодия. |
Результаты кариотипирования помогут доктору выбрать тактику ведения бесплодной пары или пациентки с привычным невынашиванием беременности, а также оценить шансы супругов на здоровое потомство.
Также в МЖЦ у вас есть возможность сдать биоматериал (кровь или слюну) для детального ХМА-анализа, диагностирующего более 1000 генетических синдромов и болезней, связанных с аберрациями.

Содержание
Генетическое исследование (определение кариотипа) нужно проводить будущим родителям и, при необходимости, плоду.
Нормальный набор хромосом
Известно, что вероятность невынашивания беременности значительно выше при хромосомных нарушениях у родителей. Поэтому данное обследование супругов применяется при привычном невынашивании беременности и бесплодии. Генетическое обследование помогает не только установить причину бесплодия, но и прогнозировать возможность рождения детей с хромосомной патологией. Поэтому большое значение придается дородовой диагностике хромосомных аберраций.
Изменения структуры хромосом
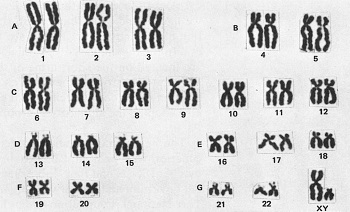 Если имеются структурные аномалии хромосомы, то в кариотипе указывается: p короткое плечо хромосомы, q — длинное плечо, t — транслокация. Например, при делеции короткого плеча хромосомы 5 женский кариотип будет выглядеть так: 46, хх, 5p- (синдром «кошачьего крика»). Мать ребёнка с синдромом Дауна, обусловленным транслокацией хромосомы 14/21, будет иметь кариотип 45, ХХ, t (14q; 21q). Измененная хромосома образуется при слиянии длинных плеч хромосомы 14 и 21, а короткие плечи теряются. В любом случае, по получению анализа необходимо обратиться к генетику, который подробно объяснит значение результатов, если в них имеются отклонения.
Если имеются структурные аномалии хромосомы, то в кариотипе указывается: p короткое плечо хромосомы, q — длинное плечо, t — транслокация. Например, при делеции короткого плеча хромосомы 5 женский кариотип будет выглядеть так: 46, хх, 5p- (синдром «кошачьего крика»). Мать ребёнка с синдромом Дауна, обусловленным транслокацией хромосомы 14/21, будет иметь кариотип 45, ХХ, t (14q; 21q). Измененная хромосома образуется при слиянии длинных плеч хромосомы 14 и 21, а короткие плечи теряются. В любом случае, по получению анализа необходимо обратиться к генетику, который подробно объяснит значение результатов, если в них имеются отклонения.
Если выявлена проблема у одного из родителей, генетик делает заключение о риске наследования ребенком того или иного заболевания или порока развития. Если беременность возможна, то все равно проводится исследование кариотипа плода, ведь не все пороки развития можно диагностировать при УЗИ, тем более, что это возможно в более поздние сроки. Определение кариотипа плода в клетках хориона дает возможность ранней диагностики наследственной патологии. В случае выявления порока развития плода, который не совместим с жизнью, проводится прерывание беременности в ранние сроки. В более поздние сроки беременности исследуются околоплодные воды и клетки кожи плода, которые получают при амнио- и кордоцентезе.
Кариотипирование плода
В обязательном порядке проводится предимплантационная генетическая диагностика при ЭКО, которая позволяет обнаружить серьезные отклонения в количестве хромосом.
Самые важные и интересные новости о лечении бесплодия и ЭКО теперь и в нашем Telegram-канале @probirka_forum Присоединяйтесь!
Микроделеция 22-й хромосомы у ребенка с задержкой физического развития: особенности эндокринного статуса, возможности терапии, клиническое наблюдение
Опубликовано в журнале:
« Практика педиатра » № 4 Ноябрь-декабрь, 2019
Е.В. Тозлиян, канд. мед. наук, педиатр-эндокринолог, генетик, ОСП «Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева» ФГБОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» МЗ РФ, г. Москва
Резюме. Представлены данные литературы, отражающие проявления, вопросы диагностики и методы лечения синдрома делеции 22-й хромосомы у детей. Подчеркнуты основные трудности при проведении дифференциальной диагностики и выявлении данного заболевания. Представлено собственное клиническое наблюдение. Показана важность междисциплинарного подхода.
Ключевые слова: дети, делеция, 22q11.2, синдром, хромосома, хромосомные заболевания, специфический фенотип, велокардиофациальный синдром.
Summary. The paper gives the data available in the literature, which reflect the manifestations, diagnosis, and current treatments syndrome of deletion chromosome 22 (22q11.2) in children. Emphasis is laid on major difficulties in the differential and true diagnosis of this disease. The author describes a clinical case. The article presents the importance of the interdisciplinary method.
Keywords: children, deletion, 22q11.2, syndrome, chromosome, chromosomal diseases, specific phenotype, velocardiofacial syndrome.
Актуальность современных молекулярно-генетических методов исследования в диагностике хромосомных заболеваний
Хромосомные болезни занимают одно из ведущих мест в структуре врожденной и наследственной патологии человека. Они обусловлены числовыми или структурными аномалиями хромосом, которые приводят к инвалидизации и часто к летальным исходам. Диагностика, в том числе пренатальная, такой частой патологии, как хромосомные синдромы, представляет большую медицинскую, научную и социальную значимость. Спектр хромосомных аномалий (ХА), являющихся причинами наследственных синдромов, широк и представлен не только числовыми аберрациями (например, наиболее распространенными трисомиями по 21-й, 13-й, 18-й хромосомам или моносомией по 10-й хромосоме), но и разнообразными структурными нарушениями хромосом, сопровождающимися дупликациями или делециями генетического материала. Значительная часть ХА представлена более мелкими аберрациями (микроделециями и микродупликациями), которые не выявляются при стандартном цитогенетическом анализе и могут быть диагностированы только при использовании современных молекулярно-цитогенетических или молекулярных методов исследования.
Перечень таких микроделеционных и микро-дупликационных хромосомных синдромов постоянно увеличивается в связи с расширением возможностей их диагностики. Частота микроделеционных синдромов среди новорожденных достаточно высокая (от 1:4000 до 1:30 000). В большинстве своем клинические проявления микроделеционных синдромов в постнатальном периоде включают задержку психомоторного и физического развития разной степени выраженности, множественные стигмы дизэмбриогенеза, пороки развития, а также различные функциональные нарушения.
Группа синдромов, обусловленных делецией 22q11.2, обозначаемая далее как синдром делеции 22q11.2 (СД22q11.2), включает несколько синдромов с различными названиями и во многом перекрывающимися клиническими признаками. Основными синдромами этой группы являются: велокардиофациальный синдром, синдром конотрункальных и лицевых аномалий и синдром Ди Джорджи. В настоящее время СД22q11.2 считается самым распространенным в популяции человека. В зарубежной литературе он обозначается как 22q11.2DS (22q11.2 deletion syndrome) [3, 5, 7, 8].
Раннее обнаружение ХА позволяет своевременно составить правильный план ведения пациента, скорректировать лечение, основываясь на понимании этиологии и патогенеза данного заболевания, и в итоге предупредить возникновение возможных осложнений. Современный уровень молекулярной диагностики, в том числе молекулярно-цитогенетическое исследование с использованием, например, методов флуоресцентной гибридизации in situ (fluorescent in situ hybridisation, FISH), сравнительной геномной гибридизации (comparative genomic hybridisation, CGH) либо мультиплексной лигандзависимой амплификации (multiplex ligand-dependent probe amplification, MLPA), позволяет верифицировать диагноз СД22q11.2 с достоверностью до 95%, что используют как в пре-, так и в постнатальном периодах [11, 12].
Составление алгоритма ведения такой категории пациентов на основании результатов молекулярной диагностики имеет существенное значение для медицинской практики, в частности практики педиатра.
Терминология
Однако единая этиология СД22q11.2 была выяснена несколько позже. В настоящее время используются названия «синдром делеции 22q11.2», или «синдром Ди Джоржи», или «велокардиофациальный синдром».
Исторически в литературе синдром часто разделяли на полный и неполный (частичный). Термин «полный синдром делеции 22-й хромосомы» использовали у пациентов со всем спектром типичных проявлений, включая выраженный иммунодефицит. Термин «частичный синдром делеции 22-й хромосомы» применяли у пациентов, которые имели лишь некоторые типичные признаки, особенно без проявлений выраженного иммунодефицита. Частичный СД22q11.2 в значительной степени превалирует по количеству по сравнению с полным [8, 10, 12].
Этиология и патогенез
Цитогенетические и молекулярные исследования показали, что делеция 22q11.2 является ведущей причиной синдрома. Анализ ДНК пациентов с СД22q11.2 выявил, что в 85-90% случаев выпадающий участок является одним и тем же. Локализация проецируется между D22S427 на 22q11.21 и D22S801 на 22q11.23. В этом участке локализовано не менее 40 генов, что составляет около 3 млн пар нуклеиновых оснований. В формировании фенотипа играет роль комплексное нарушение экспрессии и взаимодействия генов, их модификаторов и других составляющих, что приводит к дальнейшему нарушению эмбрио- и органогенеза. Соответственно при отсутствии или нарушении функции и экспрессии генов и дальнейших процессов происходит формирование пороков развития, характерных для СД22q11.2. В редких случаях синдром становится проявлением перестроек других хромосом.
Клинические проявления
Для СД22q11.2 характерен полиморфизм клинических проявлений. Те или иные клинические симптомы могут дебютировать в разном возрасте. Так, у новорожденных и детей первого года жизни преобладают ВПС и гипокальциемия, в то время как с возрастом появляются проблемы другого характера: задержка психомоторного развития, инфекционные и аутоиммунные осложнения.
Далее представлены ключевые симптомы и признаки, сочетание которых должно насторожить врача. Такому пациенту необходимо дообследование для исключения СД22q11.2.
Эндокринные нарушения. Кардиологическим расстройствам при СД22q11.2 могут сопутствовать эндокринные нарушения, отягощающие состояние пациента. Если у ребенка диагностированы конотрункальные пороки сердца, то в общем порядке следует определить содержание кальция в плазме крови, а также провести ультразвуковое исследование щитовидной и околощитовидной желез. При наличии у ребенка симптомов гипопаратиреоза следует выполнить молекулярно-генетическое исследование на наличие СД22q11.2, поскольку эта ХА является наиболее частой причиной данной патологии в детском возрасте. Диагноз гипопаратиреоза может быть упущен при наличии тяжелых врожденных конотрункальных пороков сердца. Однако если гипокальциемия не была диагностирована в первые месяцы жизни ребенка, то в дальнейшем, с замедлением роста и уменьшением потребности в кальции, симптомы гипопаратиреоза нередко нивелируются, но при этом негативно сказываются впоследствии на развитии организма и могут проявляться в виде резистентной формы эпилепсии, умственной отсталости разной степени выраженности, задержки роста и полового развития, патологии зрения. При проведении компьютерной томографии головного мозга могут встречаться кальцификаты в базальных ганглиях (синдром Фара), подтверждающие длительную гипокальциемию. Скрининг с определением содержания общего и ионизированного кальция, фосфатов крови, паратиреоидного гормона представляется необходимой составляющей алгоритма диагностики и лечения детей с СД22q11.2.
При обнаружении снижения темпов роста ребенка для исключения гипофизарной недостаточности рекомендуется исключить соматотропную недостаточность (базальные и стимуляционные тесты, рентген кистей). Гипофизарная недостаточность наблюдается у части пациентов с СД22q11.2, особенно при сопутствующих лицевых аномалиях.
Как уже было отмечено, важная роль в патогенезе нарушений развития органов и систем у плода в процессе эмбриогенеза отводится гену ТВХ1, который в том числе детерминирует органогенез щитовидной железы. Именно поэтому при СД22q11.2 необходимо проведение скрининга на гипотиреоз. У части пациентов аутоиммунный процесс может привести к гипертиреозу с повышенным уровнем антител к антигенам щитовидной железы и к риску развития болезни Грейвса или тиреоидита Хашимото. В алгоритм диагностики и лечения детей с СД22q11.2 входят определение концентрации тиреоидных гормонов и назначение гормональной терапии (препаратов левотироксина) при гипотиреозе и антитиреоидных препаратов при гипертиреозе. Таким образом, детский эндокринолог должен наблюдать пациентов с СД22q11.2 с раннего возраста и в течение последующей жизни [9, 14, 15].
Краниофациальные нарушения. Поражение носоглоточного аппарата выявлено примерно в 70% случаев и проявляется велофарингеальной недостаточностью, расщеплением нёба, губы, раздвоением уздечки нёба, гнусавым оттенком голоса, также описаны снижение обоняния, кондуктивная и/или сенсоневральная тугоухость. Особенности фенотипа (рис. 1): удлиненное лицо, микрогнатия или ретрогнатия, широкая переносица, мелкие зубы, асимметрия лица при плаче, опущенные вниз уголки рта, глазной гипертелоризм, низко посаженные и деформированные ушные раковины, бульбообразный кончик носа.
Рис. 1. Особенности фенотипа при синдроме делеции 22-й хромосомы

Иммунологические нарушения. Поскольку в 1% случаев у пациентов с полной делецией 22-й хромосомы обнаруживается тяжелый селективный Т-клеточный иммунодефицит с очень низким количеством или полным отсутствием Т-клеток, диагностика иммунных нарушений является неотъемлемой частью алгоритма ведения [16]. Нарушения Т-клеточного иммунитета при СД22q11.2 осложняют состояние ребенка, особенно на фоне тяжелых проявлений ВПС, и играют одну из определяющих ролей в восстановительном послеоперационном периоде. В алгоритм диагностики и лечения больных с СД22q11.2 входит обязательное исследование функции тимуса: ультразвуковое исследование для исключения его гипо- или аплазии; развернутый анализ крови с дифференцированным подсчетом лейкоцитов в абсолютном значении; определение концентрации иммуноглобулинов (Ig) M, A, G; фенотипирование лимфоцитов. Пациентам не показана вакцинация живыми вакцинами. Диспансерное наблюдение врачом-иммунологом ребенка с СД22q11.2 является необходимой составляющей алгоритма его ведения.
Психические расстройства. Дети с СД22q11.2 составляют группу риска по развитию психиатрических заболеваний [17]. Расстройства в эмоциональной и поведенческой сферах в раннем возрасте могут быть малозаметными, но носить прогрессирующий характер. Распространенность психических заболеваний у пациентов с СД22q11.2 составляет от 60 до 93%. Проявления нарушений со стороны психоэмоциональной сферы у этих пациентов следующие: синдром дефицита внимания с гиперактивностью, расстройства аутистического спектра, генерализованное тревожное расстройство, специфическая фобия, депрессия, шизофрения. Ранняя психолого-педагогическая поддержка семьи ребенка с СД22q11.2 способствует своевременному созданию специальных развивающих условий для реализации его психического потенциала и предупреждает тяжелые необратимые нарушения в его психоэмоциональной сфере. Наблюдение психологом и психиатром детей с СД22q11.2 является важной составляющей их ведения.
Нарушения психомоторного и речевого развития. У детей с СД22q11.2 отмечается нарушение мелкой моторики вследствие снижения мышечного тонуса, в связи с чем дети могут испытывать трудности с четким выполнением быстрых движений. Задержка речевого развития при наличии врожденной расщелины у ребенка с СД22q11.2 предполагает обязательное логопедическое обучение в до- и послеоперационном периодах при устранении дефекта нёба. В образовательной сфере следует обратить внимание на трудности в обучении ребенка и на его способности, чтобы выбрать оптимальную программу подготовки. Составление индивидуальной программы обучения ребенка с СД22q11.2 способствует предотвращению фрустраций, неуверенности в себе и отсутствия мотивации к учебе [18]. Таким образом, алгоритм диагностики и лечения СД22q11.2 предполагает обязательное выявление нарушений коммуникативных, поведенческих и когнитивных функций, комплексную медицинскую, психологическую и педагогическую поддержку детей уже с раннего возраста, составление индивидуальных программ обучения с учетом способностей ребенка.
Нарушения других органов и систем. У детей с СД22q11.2 могут также встречаться нарушения со стороны других органов и систем: мальротация кишечника, атрезия пищевода, трахеопищеводный свищ, атрезия ануса, болезнь Гиршпрунга, одно- или двусторонняя почечная агенезия, дисплазия почки, поликистоз почек, гидронефроз, гипоспадия, крипторхизм, паховая и пупочная грыжи.
Алгоритм диагностики и лечения СД22q11.2 предполагает применение мультидисциплинарного комплексного подхода и слаженное взаимодействие специалистов различных направлений.
Диагностика
Диагноз подтверждают с помощью молекулярно-генетического исследования: положительный тест методом флуоресцентной гибридизации in situ (FISH) с ДНК-зондом TUPLE 1 (HIRA); обнаружение локусов с отсутствием генетического материала методом полимеразной цепной реакции и электрофореза с помощью анализа микросателлитного полиморфизма по определенным локусам, перекрывающим область делеции 22-й хромосомы; мутации в гене ТВХ1.
Лабораторная диагностика:
Синдром делеции 22q11.2 встречается спорадически более чем в 90% случаев. У 10% пациентов делеция наследуется от одного из родителей. Наличие родителя с СД22q11.2 считается фактором риска, так как наследование является аутосомно-доминантным, следовательно, 50% вероятности того, что патология унаследуется. При этом обнаружено, что у потомства синдром более выражен, чем у родителя. Пренатальная диагностика необходима, если один из родителей имеет СД22q11.2, так как в 50% случаев патология наследуется с более выраженной тяжестью.
Лечение
Алгоритм ведения детей с СД22q11.2 позволяет своевременно обнаружить нарушения органов и систем, характерные для этой ХА, и скорректировать план лечения ребенка. Мультидисциплинарный подход к ведению детей с такой патологией является «золотым стандартом», поскольку ускоряет диагностику, повышает эффективность лечения, сокращает сроки реабилитации и тем самым улучшает качество жизни ребенка. Эффективность реабилитации зависит от правильной координации действий врачей различных специальностей.
Клиническое наблюдение
Девочка К., 2 года. Родители обратились за консультацией в Московский областной клинико-диагностический центр для детей с жалобами на задержку физического и психомоторного развития, мышечную гипотонию, запоры, плохой (избирательный) аппетит, нарушение глотания (предпочитает протертый стол).
На фоне медикаментозной терапии левотироксином улучшились темпы роста, увеличился словарный запас, появилась фразовая речь, ребенок стал активнее, улучшился аппетит, нормализовался стул. В общеклиническом анализе крови уровень гемоглобина приблизился к норме.
Назначен комплекс реабилитационных мероприятий, направленных на стимуляцию психомоторного развития: массаж, физиолечение, лечебная физкультура. Девочка постоянно получает курсы физиолечения, массажа, работает инструктор по плаванию, что эффективно сказывается на ее моторном развитии.
Неврологом назначена ноотропная терапия по схеме.
Динамика развития ребенка позволяет прийти к заключению: назначение комплексных реабилитационных мероприятий может способствовать эффективному физическому и психомоторному развитию. Только совместные усилия врачей различных специальностей помогают детям с врожденной и наследственной патологией социализироваться в обществе, мало отличаться от окружающих сверстников и быть счастливыми вместе со своими родителями и близкими.
Заключение
Хромосомные болезни занимают одно из ведущих мест в структуре врожденной и наследственной патологии человека. Диагностика, в том числе пренатальная, такой частой патологии, как хромосомные синдромы, представляет большую медицинскую, научную и социальную значимость. Установление диагноза больному ребенку позволяет семье определить наличие или отсутствие наследственной патологии и получить не только аргументированную медико-генетическую консультацию по поводу прогноза потомства, но и рекомендации по пренатальной диагностике конкретного заболевания. Это в свою очередь может стимулировать родителей к новой беременности, которая будет протекать при гарантированном благоприятном прогнозе здоровья будущего ребенка.
Кариотипирование супругов. Анализ на кариотип
Кариотип и кариотипирование супругов
Кариотипирование супругов – это углубленное лабораторное обследование для выявления изменений кариотипа, которое сдает пара в тех случаях, когда необходимо понять причину бесплодия, невынашивания беременности, а также заранее исключить генетические проблемы перед протоколом ЭКО и планированием беременности.
Кариотип – хромосомный набор человека с совокупностью признаков. Генетический фактор занимает достаточно большой процент среди супружеских пар с бесплодием, невынашиванием беременности, а также в группах мужчин с тяжелым нарушением сперматогенеза.
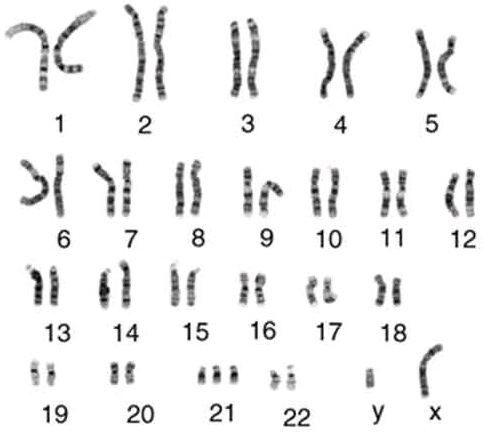
Анализ на кариотип сдают один раз в жизни. Поскольку это важный генетический анализ, то рекомендуется сдавать в специализированных центрах. В нашей лаборатории кариотипирование проводят высококвалифицированные лабораторные генетики. Анализируется материал всех 23 пар хромосом. Результат выдается согласно международной цитогенетической номенклатуре.
В норме результаты кариотипа выглядят следующим образом:
46, XX – нормальный женский кариотип;
46, XY – нормальный мужской кариотип.
Изменения кариотипа могут представлять собой изменения количества хромосом (анеуплоидии) либо структуры хромосом (или аберрации: транслокации, инверсии и др.). Внешне здоровый человек может быть носителем хромосомных аномалий, которые могут являться причиной бесплодия, невынашивания беременности или рождения у супружеских пар детей с пороками развития.

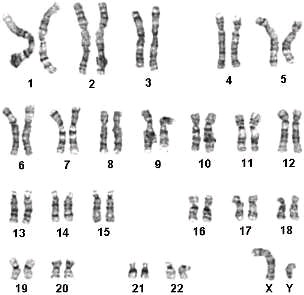
46, XX – нормальный женский кариотип 46, XY – нормальный мужской кариотип
46, XY, +21 – дополнительная хромосома 21 (синдром Дауна)
Классическим методом определения кариотипа является цитогенетический. Также этим методом выполняют кариотипирование супругов. Метод основан на культивировании клеток крови с последующим приготовлением и фотографированием препаратов окрашенных хромосом. Метод ХМА представляет собой современную молекулярную технологию исследования кариотипа и показан при задержке развития и роста человека, наличием врожденных пороков развития (ВПР) у детей, аутизме, подозрении на микроделеционные синдромы.
В Лаборатории ЦИР кариотипирование супругов проводят цитогенетическим методом:
Что такое аберрации?
Почему экспертного уровня?
Мы являемся одной из немногих клиник, работающих более 20 лет в области репродукции. В нашей лаборатории цитогенетики кариотипирование проводится специалистами высокого уровня, где идет просмотр каждой хромосомы.
Как сдать анализ на кариотип. Подготовка.
Смотрите также:
«Замершая» беременность: каковы причины? Какое значение имеет генетический фактор в развитии «замершей» беременности?
Анализ на кариотип (кариотипирование). Как интерпретировать анализ на кариотип? Отвечает Гузов И.И.
ГЕНЫ и БЕРЕМЕННОСТЬ. Генетические причины невынашивания беременности.
Мозаичность кариотипа
Выявление мозаицизма кариотипа зависит от процентного содержания его (если есть) в кариотипе и от выбранной методики анализа. Однозначно при ХМА более вероятно обнаружение мозаицизма в пределах разрешающей способности (наличие более 25% в кариотипе). Также можно обнаружить мозаицизм при наличии его в исследуемых клетках в Определении кариотипа с аберрациями цитогенетическим методом. В связи с ограничениями цитогенетического метода если процент мозаицизма мал, то, скорее всего, его можно не увидеть.
Хромосомный микроматричный анализ (ХМА)
Однако для диагностики ряда заболеваний, связанных с хромосомными аномалиями, существует более современная технология исследования кариотипа – хромосомный микроматричный анализ. Анализ на кариотип выполняется молекулярно-генетическим методом aCGH (микроматричная сравнительная геномная гибридизация), который в отличие от классического цитогенетического метода, имеет высокую разрешающую способность, позволяющую обнаружить более мелкие структурные изменения кариотипа.
Методы диагностики хромосомной патологии