Болезнь Аддисона

Болезнь Аддисона (гипокортицизм, хроническая надпочечниковая недостаточность) — это редкое заболевание, характеризующееся недостаточной выработкой надпочечниками глюкокортикоидного гормона кортизола и минералокортикоидного гормона альдостерона.
Чаще всего гипокортизицм, которому подвержены как мужчины, так и женщины, возникает в 30-50 лет, однако дебют (первое клиническое проявление) заболевания может наблюдаться в любом возрасте.
Распространенность болезни Аддисона во всем мире составляет 40-60 пациентов с установленным диагнозом БА на 1 миллион человек.
Причины возникновения
Как правило, болезнь Аддисона возникает из-за повреждения или разрушения коры надпочечников в результате развития аутоиммунного процесса (иммунная система воспринимает кору надпочечников как чужеродную, атакует и разрушает ее) или другого аутоиммунного заболевания, имеющегося у пациента.
К другим причинам возникновения заболевания относятся:
Дефицит глюкокортикоидных и минералокортикоидных гормонов, вызванный снижением функции коры надпочечников, называют первичной надпочечниковой недостаточностью. Если надпочечники не способны продуцировать эти гормоны по другим причинам (например, из-за аномалий гипофиза или его дисфункции), речь идет о вторичной надпочечниковой недостаточности.
Симптомы
Болезнь Аддисона прогрессирует медленно, в течение нескольких месяцев. Симптомы могут варьироваться от человека к человеку. Первые клинические проявления часто неспецифичны, то есть сходны с симптомами других — распространенных — заболеваний.
Обычно на начальных стадиях заболевания пациент не ощущает никаких симптомов. К сожалению, дебют БА ассоциирован с 90% повреждения коры надпочечников.
К симптомам болезни Аддисона относятся:
Иногда болезнь Аддисона, не проявлявшая себя ранее, может возникнуть внезапно и остро — например, при отсутствии необходимого лечения или в результате воздействия стрессовых факторов (несчастный случай, травма, хирургическое вмешательство), инфекции, другого заболевания. В этом случае речь идет о жизнеугрожающем состоянии — т. н. аддисоническом кризе или острой надпочечниковой недостаточности. Характерные признаки: сильная слабость, спутанность сознания, боли в спине, ногах и животе, рвота и диарея, низкое артериальное давление, гиперкалиемия, гипонатриемия.
Диагностика
Диагноз устанавливается на основании анамнеза, жалоб и симптомов пациента, результатов лабораторной и инструментальной диагностики (КТ, МРТ).
Анализы крови направлены на определение уровня натрия, калия, кортизола и адренокортикотропного гормона (АКТГ), который стимулирует выработку гормонов корой надпочечников, а также на выявление специфических антител, ассоциированных с болезнью Аддисона.
Пациенту может быть назначена стимуляционная проба АКТГ, во время которой (для стимуляции выработки кортизола) вводится адренокортикотропный гормон. Если стимуляция не приводит к выработке нормального уровня кортизола, это указывает на повреждение надпочечников или нарушение их функции.
Также проводится инсулин-индуцированная гипогликемическая проба, которая помогает установить взаимосвязь характерных симптомов болезни Аддисона с аномалиями гипофиза или нарушениями его функции. Во время этого теста пациенту вводится быстродействующий инсулин, уровень сахара в крови замеряется до и после инъекции.
Для оценки функции надпочечников и их структурных особенностей дополнительно проводится визуализация с помощью КТ и МРТ. Эти исследования помогают уточнить характерные изменения, наблюдаемые при развитии болезни Аддисона.
Дифференциальный диагноз
Чаще всего болезнь Аддисона дифференцируют (различают) со следующими заболеваниями:
Лечение болезни Аддисона
Лечение болезни Аддисона основано на применении медикаментозной терапии, направленной на замещение глюкокортикоидного и минералокортикоидного гормонов. Рекомендуется пероральный прием гидрокортизона и флудрокортизона, дозировка препаратов подбирается индивидуально. Терапия проводится на протяжении всей жизни.
Некоторым пациентам может быть предложена андроген-заместительная терапия (тестостерон или андростендион). Больные, получающие такое лечение, сообщают о снижении проявлений усталости, улучшении настроения и психоэмоционального состояния в целом, повышении либидо (у женщин), однако эти подходы к лечению пока исследуются.
Лечение аддисонического криза включает внутривенное введение гидрокортизона, жидкостей, электролитов и препаратов, нормализующих артериальное давление.
Особенности и преимущества лечения болезни Аддисона в клинике Рассвет
Своевременная диагностика и эффективное лечение редких синдромов и болезней входят в область интересов всех врачей клиники Рассвет.
Наши эндокринологи — врачи высокой квалификации, прекрасно подготовлены и имеют большой практический опыт.
Пациентам с подозрением на болезнь Аддисона проводятся все необходимые диагностические исследования для подтверждения или исключения диагноза. При необходимости индивидуально подбирается адекватная схема терапии.
Рекомендации врача-эндокринолога клиники Рассвет пациентам с болезнью Аддисона
Помимо приема препаратов заместительной терапии, пациенту необходимо следить за тем, чтобы получать достаточное количество соли (натрия) из рациона, особенно во время физических нагрузок, при жаре или желудочно-кишечных расстройствах.
Дозировка принимаемых лекарств может быть временно увеличена, если пациент испытывает стресс, связанный с проведением операции, инфекционными и другими заболеваниями. Если из-за тошноты и рвоты пациент не может принимать необходимые препараты перорально, ему назначаются инъекции.
Пациентам с болезнью Аддисона важно знать, что прием стероидов в их случае не связан с серьезными побочными эффектами, поскольку осуществляется восполнение недостатка выработки гормонов. При правильном подборе дозировки лекарств побочные эффекты практически не наблюдаются.
Пациентам с болезнью Аддисона необходимо постоянно иметь при себе медицинский документ, свидетельствующий о наличии заболевания, для случаев оказания экстренной медицинской помощи.
Болезнь Аддисона нельзя предотвратить, но для нее существует эффективное лечение. При своевременной диагностике, правильном подборе терапии, соблюдении мер профилактики острой надпочечниковой недостаточности, ожидаемая продолжительность жизни у пациентов с этим заболеванием не снижается.
Синдром Шегрена – что это такое? Причины, симптомы и лечение у опытных врачей медицинской Клиники МЕДСИ
Оглавление
Синдром Шегрена – аутоиммунное системное поражение соединительной ткани. Патология отличается тем, что в нее вовлечены железы внешней секреции (преимущественно слезные и слюнные). Вследствие развития заболевания появляется выраженная сухость кожи, носоглотки, глаз, рта, трахеи и влагалища. Также сокращается выработка пищеварительных ферментов. Патология может развиваться как самостоятельная или сопровождать склеродермию, дерматомиозит и другие заболевания. Лечение симптома Шегрена следует начинать после обнаружения первых же признаков.
Патоморфология
На раннем этапе в процесс вовлекаются мелкие протоки желез. При развитии заболевания железистая ткань атрофируется и замещается соединительной. Это приводит к нарушению функций пораженного органа. Нередко даже при отсутствии других выраженных симптомов синдрома Шегрена у пациентов отмечаются признаки воспаления слюнных желез.
Причины развития
Причины возникновения патологии в настоящий момент до конца не установлены.
Наиболее вероятной считают теорию о патологической реакции иммунной системы, которая развивается в ответ на повреждение клеток ретровирусом (герпесом, ВИЧ и др.). Как вирусы, так и клетки эпителия, измененные под их воздействием, воспринимаются иммунной системой человека как чужеродные. Иммунная система защищает организм и вырабатывает антитела. Это и приводит к разрушению тканей железы. Нередко синдром Шегрена передается по наследству, встречается у родителей и детей, у близнецов.
Спровоцировать развитие патологии могут следующие факторы (нередко их комбинации):
Симптомы синдрома Шегрена
Симптомы синдрома Шегрена во много зависят от причин заболевания, но всегда требуют устранения (лечения), так как существенно снижают уровень качества жизни пациента.
К основным железистым признакам относят:
К внежелезистым проявлениям патологии относят:
Нередко пациенты жалуются на повышенную чувствительность к ряду медикаментозных препаратов (нестероидным противовоспалительным средствам, антибиотикам и др.).
Если у вас появилась сыпь на теле, повысилась температура, пересыхают слизистые или обнаруживаются другие симптомы, и вы не знаете, что это, но хотите начать лечение, следует обратиться к специалисту: только он может провести диагностику и выявить синдром Шегрена или другую патологию.
Диагностика
Такие симптомы, как жжение и сухость глаз, например, не всегда свидетельствуют о синдроме Шегрена, но становятся причиной обращения к врачу с целью профилактики и лечения. Профессионалу очень важно точно распознать заболевание.
Диагностировать синдром Шегрена можно при наличии воспалительного процесса. Но в некоторых случаях воспаление провоцируется другими патологиями (например, сахарным диабетом). Для этого заболевания также характерно снижение секреции слюны. По этой причине диагностика должна быть максимально точной.
Наиболее информативным методом является биопсия слюнных и слезных желез с последующей гистологией полученного материала. Она проводится быстро и не доставляет пациентам выраженного дискомфорта. Фрагменты слизистых оболочек исследуются под микроскопом. Благодаря этому специалистам удается зафиксировать поражение желез.
Осложнения
К основным осложнениям синдрома Шегрена относят:
Патология прогрессирует как без лечения, так и в том случае, если терапия проводится неправильно. Именно поэтому следует обращаться только к высококвалифицированным врачам, располагающим опытом работы с пациентами с синдромом Шегрена.
Лечение заболевания
Основными задачами в терапии синдрома Шегрена являются снятие воспаления пораженных органов и устранение симптомов слизистых оболочек.
Для устранения воспалительного процесса назначаются:
В ходе подготовки к лечению пациентам проводят плазмаферез, позволяющий очистить кровь.
Для профилактики сухости конъюнктивы назначают препараты искусственной слезы и мази. Уход за полостью рта заключается в тщательном полоскании после каждого приема пищи.
Важно! Лечение синдрома Шегрена всегда проводится только под контролем врача-ревматолога.
Прогноз
Синдром Шегрена опасен тем, что может приводить к повреждению жизненно важных органов, постепенно прогрессировать. Бывают и случаи длительных ремиссий, когда патология никак не проявляет себя и больному кажется, что он полностью излечился, но болезнь внезапно возвращается. Одним пациентам помогает только симптоматическое лечение, другие долгое время борются с постоянным дискомфортом. Качество жизни многих больных существенно снижается. Пациенты страдают от суставных болей, сухости слизистых, упадка сил.
Важно! Пациенты с синдромом Шегрена подвержены высокому риску неходжкинской лимфомы. У некоторых больных развиваются другие онкологические заболевания, которые могут стать причиной не только снижения качества жизни, но и смерти.
При правильном и комплексном лечении пациенты могут рассчитывать на длительную и стойкую ремиссию. Но терапия должна быть комплексной и начаться как можно раньше – после обнаружения первых же признаков патологии.
Преимущества лечения синдрома Шегрена в МЕДСИ
Если вы хотите записаться на прием к ревматологу, позвоните
Болезнь сандхоффа что это такое
а) Терминология:
1. Сокращения:
• Ганглиозидоз (GM2)
2. Синонимы:
• Болезнь Тея-Сакса (БТС), болезнь Сандхоффа (БС)
3. Определение:
• Наследственная лизосомная болезнь накопления:
о Характеризуется накоплением GM2-ганглиозидов в головном мозге
• Выделяют три биохимически различных, но клинически неотличимых типа заболевания:
о БТС
о БС
о GM2, вариант АВ (редко)
• Существуют инфантильная, ювенильная и взрослая формы БТС и БС
• GM2, вариант АВ существует только в детской форме
1. Общие характеристики ганглиозидоза:
• Лучший диагностический критерий:
о Инфантильная форма:
— Гипоинтенсивные на Т2-ВИ, гиперинтенсивные на Т1-ВИ (гиперденсные при КТ) таламусы
— Слегка гиперинтенсивное на Т2-ВИ полосатое тело
о Ювенильная/взрослая форма:
— Атрофия мозжечка
• Локализация:
о Инфантильная форма: таламусы, полосатое тело, БВ больших полушарий > > БВ мозжечка:
— Мозолистое тело (МТ) сохранно
о Ювенильная/взрослая форма: мозжечок, БВ больших полушарий:
— Редкое вовлечение ствола мозга (по типу объемного образования) и мозолистого тела
• Морфология:
о Симметричное вовлечение глубокого серого вещества
о Поздняя стадия заболевания: атрофия
2. КТ признаки ганглиозидоза:
• Бесконтрастная КТ:
о Инфантильная форма:
— Гиперденсные таламусы (классический, но вариабельный признак)
— Гиподенсные полосатое тело, БВ
о Ювенильная/взрослая форма
— Атрофия мозжечка:
— ± гиподенсное БВ больших полушарий
• КТ с контрастированием:
о Патологическое накопление контраста отсутствует
4. УЗИ:
• Инфантильная форма: гиперэхогенные таламусы
5. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ (КТ может подтвердить изменения со стороны таламусов)
в) Дифференциальная диагностика ганглиозидоза:
1. Болезнь Краббе:
• Гиперденсные таламусы, хвостатые и зубчатые ядра
• Повышение интенсивности сигнала от БВ большого мозга, мозжечка
• Вовлечение МТ
2. Ювенильный ганглиозидоз GM1:
• Визуализационные признаки идентичны БС
• Накопление GM1-ганглиозида в головном мозге и внутренних органах
3. Нейрональный цероидный липофусциноз:
• Гиперденсные, гиперинтенсивные на Т2-ВИ таламусы, бледные шары
• Атрофия большого мозга, мозжечка
4. Мраморное состояние (status marmoratus):
• Гиперденсные, атрофичные таламусы
• Атрофия скорлупы, периролиновой области
• Тяжелая перинатальная ишемия в анамнезе
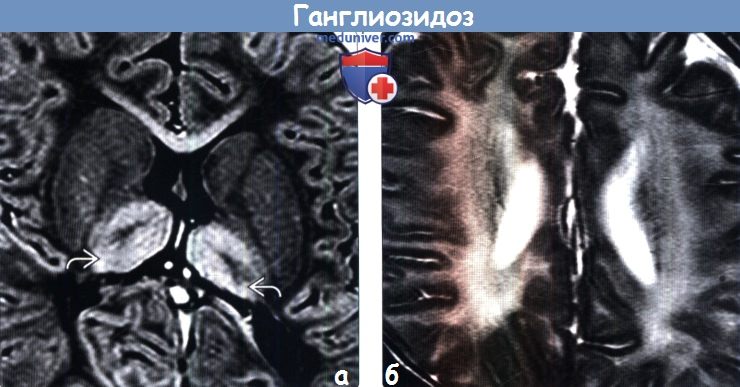 (а) МРТ, Т1-ВИ, аксиальный срез: у двухлетнего ребенка с инфантильной формой болезни Сандхоффа определяется симметричное повышение интенсивности сигнала от таламусовS Центральные гипоинтенсивные участки нетипичны. Нa фоне сохранности мозолистого тела отмечается выраженное снижение интенсивности сигнала от БВ больших полушарий.
(а) МРТ, Т1-ВИ, аксиальный срез: у двухлетнего ребенка с инфантильной формой болезни Сандхоффа определяется симметричное повышение интенсивности сигнала от таламусовS Центральные гипоинтенсивные участки нетипичны. Нa фоне сохранности мозолистого тела отмечается выраженное снижение интенсивности сигнала от БВ больших полушарий.
(б) МРТ, Т2-ВИ, аксиальный срез: у того же пациента определяется диффузное повышение интенсивности сигнала от БВ, подтверждающее распространенную гипо-/демиелинизацию. Кора больших полушарий головного мозга не изменена.
1. Общие характеристики ганглиозидоза:
• Основные патофизиологические аспекты:
о Накопление GМ2-ганглиозида в нейронах, вызванное дефицитом лизосомального фермента β-гексозаминидазы А
• Эмбриология-анатомия:
о GМ2-ганглиозид локализуется в мембранах нейронов; играет роль в распознавании других клеток, синаптогенезе
о β-гексозаминидаза А (НехА) и GМ2-активаторный белок (GMAP) необходимы для катаболизма лизосомального GM2-ганглиозидоза
о НехА представляет собой 1 из 3-х изоферментов β-гексозаминидазы, сформированной посредством димеризации α- и β-субъединиц
— НехА = αβ димер, НехВ = ββ, HexS = αα
о НехА и НехВ являются основными формами; HexS-небольшая форма с неясной физиологической функцией
о НЕХА, локус 15q23—24, кодирует α-субъединицу о НЕХВ, локус 5q13, кодирует β-субъединицу
о GM2A, локус 5q31.3-q33.1, кодирует GMAP
• Генетика:
о Аутосомно-рецессивное наследование о > 100 различных мутаций НЕХА вызывают БТС
о > 30 различных мутаций НЕХВ вызывают БС
о 4 мутаций GM2A вызывают GM2, вариант АВ
о Мутации, допускающие остаточную активность НехА (0,5-4% от нормальной активности), наблюдаются при более легких юве-нильных/взрослых фенотипах
• Этиология:
о Накопление GМ2-ганглиозида в лизосомах нейронов вызывает деградацию, апоптоз нейронов с развитием вторичной гипо-/демиелинизации
— Накопление GМ2-ганглиозида в миелиновой оболочке может также способствовать демиелинизации
о Точный механизм апоптоза нейронов вследствие накопления GМ2-ганглиозида неизвестен:
— Активация микроглии, макрофагов и астроцитов предлагает воспалительный компонент
— Идентификация аутоантител в мышиных моделях БС предполагает аутоиммунный компонент
2. Макроскопические и хирургические особенности:
• Инфантильная форма: мегаленцефалия в раннюю стадию, атрофия в позднюю стадию:
о Желатинозная дегенерация ± кавитационные изменения БВ больших полушарий
• Ювенильная/взрослая форма: атрофия мозжечка
3. Микроскопия:
• Накопление GМ2-ганглиозидов в нейронах большого мозга
• Менее выраженное накопление GМ2-ганглиозида в глии, клетках Пуркинье, передних рогах спинного мозга, ганглионарных клетках
• ЕМ: GМ2-ганглиозид, содержащийся в мембранозных цитоплазматических телах (МЦТ) в цитоплазме нейронов, проксимальных отделах их отростков, аксонов:
о МЦТ в цитоплазме вызывают искажение и баллонную дистрофию нейронов
о МЦТ в проксимальных отделах отростков нейронов образуют меганевриты
• Гипомиелинизация, демиелинизация, валлерова дегенерация
• Ювенильная/взрослая форма GM2: накопление ганглиозидов в клетках передних рогов спинного мозга, нейронах мозжечка, базальных ганглиях, стволе мозга
о МЦТ иногда могут отсутствовать
• БС: дополнительное накопление GM2 (и глобозида) во внутренних органах
д) Клиническая картина:
1. Проявления ганглиозидоза:
• Наиболее частые признаки/симптомы:
о Инфантильная форма:
— Задержка/регрессия психомоторного развития
о Ювенильная/взрослая форма:
— Атипичная спиноцеребеллярная атаксия
о Другие признаки/симптомы:
— Инфантильный: макрокрания, гипотония, судороги, слепота (у 90% пациентов наблюдается вишнево-красное пятно в области желтого пятна), оживленный старт-рефлекс на звуковой раздражитель
— Ювенильная/взрослая форма: дизартрия, экстрапирамид-ные и пирамидные нарушения, периферическая невропатия, заикание, психоз/депрессия (на поздних стадиях, 30%)
• Клинический профиль:
о Диагноз: дефицит НехА в лейкоцитах сыворотки крови, культивированных фибробластах кожи, амниотической жидкости или образце ворсинок хориона
о Соответствующие изменения должны сопровождаться анализом ДНК с целью выявления мутации и/или исключения аллели псевдодефицита
4. Лечение:
• Поддерживающая терапия, купирование судорожного синдрома
• Новые перспективные методы лечения: ингибирование субстрата, заместительная терапия, трансплантация костного мозга, генная терапия, фармакологическая шапероновая терапия
е) Список литературы:
Редактор: Искандер Милевски. Дата публикации: 22.4.2019
Ганглиозидоз GM2 тип II (болезнь Сандхоффа, амавротическая идиотия Сандхоффа)
Что провоцирует / Причины Ганглиозидоза GM2 типа II (болезни Сандхоффа, амавротической идиотии Сандхоффа):
Болезнь Сандхоффа (GM2-ганглиозидоз, типа III, вариант 0) развивается в результате частичного дефицита гексозаминидазы А.
Патогенез (что происходит?) во время Ганглиозидоза GM2 типа II (болезни Сандхоффа, амавротической идиотии Сандхоффа):
Характерно набухание нейронов с концентрическими слоями цитоплазматических включений, но при типе II выше концентрация аспалоганглиозида GM2 в нервной ткани, а также гликосфинголипида в мезенхимальных тканях внутренних органов, что проявляется незначительной гепатомегалией.
Симптомы Ганглиозидоза GM2 типа II (болезни Сандхоффа, амавротической идиотии Сандхоффа):
К каким докторам следует обращаться если у Вас Ганглиозидоз GM2 тип II (болезнь Сандхоффа, амавротическая идиотия Сандхоффа):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Ганглиозидоза GM2 типа II (болезни Сандхоффа, амавротической идиотии Сандхоффа), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Euro lab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Euro lab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Euro lab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
СОДЕРЖАНИЕ
Симптомы и признаки
Две другие формы болезни Сандхоффа имеют похожие симптомы, но в меньшей степени. Взрослые и юношеские формы болезни Сандхоффа встречаются реже, чем младенческая. В этих случаях жертвы страдают когнитивными нарушениями (умственной отсталостью) и потерей мышечной координации, что ухудшает и в конечном итоге лишает их способности ходить; также появляются характерные красные пятна на сетчатке. Однако взрослая форма болезни иногда протекает легче и может привести только к мышечной слабости, которая ухудшает ходьбу или способность вставать с постели.
Причины
С помощью рестрикционных ферментов было обнаружено, что мутация на хромосоме 5, в частности в аллеле C1214T, вызвала у взрослых начальную форму болезни Сандхоффа. У пациента с симптомами инфантильной или ювенильной формы имеется мутация экзона I207V от отца и делеция из 16 пар оснований от матери, которая может располагаться на пяти экзонах, экзонах 1–5. 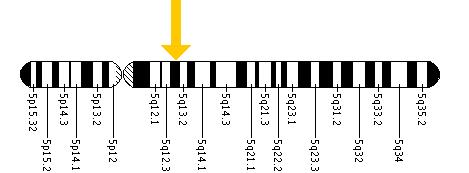
Мутации и полиморфизм
Статьи о частотах заболевания Сандхоффа среди различных групп людей содержат несоответствия друг с другом. Сообщалось о более чем 25 мутациях, помимо новых.
В одной статье говорится, что болезнь Сандхоффа обычно встречается у людей нееврейского происхождения.
Другие говорят, что это чаще встречается в:
Обнаружение нескольких мутаций у евреев-ашкенази может отражать предвзятость установления, а не высокую частоту популяций, потому что евреи-ашкенази были целевой группой в программе массового обследования на болезнь Тай-Сакса. Несколько редких мутаций SD были обнаружены, когда исследователи разрешили случаи дефицита фермента у предполагаемых носителей TSD, но ни о каких случаях самого заболевания не сообщалось.
Однако, поскольку это аутосомно-рецессивное заболевание, оно, вероятно, обнаруживается в любой этнической группе, переходящей из поколения в поколение через носителей, не проявляясь в их потомстве. Даже если в семье может не быть в анамнезе болезни Сандхоффа, у двух человек может родиться ребенок с этой болезнью. Поскольку болезнь Сандхоффа была обнаружена только в 1968 году, в течение многих лет болезнь оставалась незамеченной из-за ошибочных диагнозов.
Патофизиология
В результате прогрессирующее повреждение, вызванное накоплением ганглиозида GM2, приводит к разрушению нервных клеток, вызывая признаки и симптомы, связанные с болезнью Сандхоффа.
Диагностика
Существует три типа болезни Сандхоффа: классический детский, юношеский и взрослый с поздним началом. Каждая форма классифицируется по степени тяжести симптомов, а также по возрасту, в котором у пациента проявляются эти симптомы.
Юношеские и взрослые формы болезни Сандхоффа очень редки. Признаки и симптомы могут проявляться в детстве, подростковом или взрослом возрасте и обычно более легкие, чем те, которые наблюдаются при младенческой форме болезни Сандхоффа. Как и в младенческой форме, страдают умственные способности и координация. Характерные черты включают мышечную слабость, потерю мышечной координации ( атаксию ) и другие проблемы с движением, проблемы с речью и психические заболевания. Эти признаки и симптомы широко различаются среди людей с поздними формами болезни Сандхоффа.
В настоящее время болезнь Сандхоффа не имеет стандартного лечения и лечения. Однако человеку, страдающему этим заболеванием, необходимо правильное питание, гидратация и поддержание чистоты дыхательных путей. Чтобы уменьшить некоторые симптомы, которые могут возникнуть при болезни Сандхоффа, пациент может принимать противосудорожные препараты для лечения припадков или лекарства для лечения респираторных инфекций, а также придерживаться точной диеты, состоящей из продуктов-пюре из-за трудностей с глотанием. Младенцы с этим заболеванием обычно умирают в возрасте 3 лет из-за респираторных инфекций. Пациент должен находиться под постоянным наблюдением, потому что он может страдать от аспирации или не иметь возможности перейти от прохода к легким, а не к желудку, и их слюна попадает в легкие, вызывая бронхопневмонию. Пациент также не может кашлять и поэтому должен пройти курс лечения, чтобы встряхнуть свое тело, чтобы удалить слизь со слизистой оболочки легких. Пациентам также назначают лекарства для уменьшения симптомов, включая судороги.
История

Молекулярный дефект при болезни Сандхоффа был обнаружен, когда Конрад Сандхофф изучал биохимию сфинголипидов и ганглиозидов в лаборатории профессора Хорста Яцкевица (1912–2002), немецкого биохимика (Институт психиатрии Макса Планка, Мюнхен). В октябре 1966 года он получил замороженный материал вскрытия ребенка, страдающего амавротическим идиотизмом. Анализ гликолипидов вскоре показал отличия от всех ранее изученных случаев. Помимо накопления GM2 в нейронах, накопление GA2 было гораздо более выраженным, и в отличие от всех изученных до сих пор случаев болезни Тея-Сакса, глобозид накапливался во внутренних органах и, что наиболее важно, практически полностью отсутствовала активность гексозаминидазы. Болезнь, вызывающая дефицит катаболических ферментов гексозаминидаз, была продемонстрирована с четырьмя различными субстратами (п-нитрофенил-β-DN-ацетилглюкозаминидом, п-нитрофенил-β-DN-ацетилгалактозамидом, гликолипидом [3H] GA2 и [3H] глобозидом) в четырех разных органах. и опубликовано в 1968 году.
Смотрите также
использованная литература
Эта статья включает в себя текст из общественного достояния из Национальной медицинской библиотеки США.