Болезнь Помпе
Почему возникает болезнь Помпе?
Ген, в котором происходит мутация, кодирует белок кислую мальтазу (известна также как альфа-глюкозидаза). Это фермент необходим для расщепления полисахарида гликогена до глюкозы.
Формы заболевания
Болезнь Помпе может возникать в любом возрасте. В зависимости от того, когда появились симптомы заболевания, различают несколько его форм.
Болезнь Помпе с поздним началом. Симптомы проявляются в любом возрасте старше одного года.
Симптомы младенческой формы заболевания
О возможном развитии болезни у детей первого года жизни может свидетельствовать комплекс симптомов «вялого ребенка», который становится заметен в первые месяцы жизни. Признаки классической младенческой формы заболевания 1,3 :
Симптомы болезни Помпе с поздним началом
Иная клиническая картина возникает при болезни Помпе с поздним началом. Нередко первыми признаками становятся слабость мышц конечностей и параспинальных мышц. Обычно болезнь прогрессирует медленно, часто — на протяжении многих лет. Классические симптомы болезни Помпе с поздним началом 2 :
Лабораторно – повышение уровня креатинфосфокиназы в крови до 2000 ед. Поскольку симптомы болезни Помпе с поздним началом могут встречаться при многих других патологиях, а также из-за низкой распространенности этого заболевания, его диагностика довольно сложна. Чтобы ускорить процесс установки диагноза и как можно быстрее начать лечение, важно при появлении признаков болезни, в частности, прогрессирующей мышечной слабости, не откладывая, обратиться к неврологу или нервно-мышечному специалисту.
Диагностика и лечение болезни Помпе
Диагностика болезни Помпе проста 4 :
Болезнь Помпе
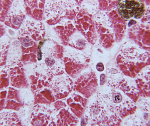
Болезнь Помпе – редкая наследственная патология, одна из форм лизосомных болезней накопления, характеризующаяся нарушением процессов расщепления гликогена в нервных и мышечных клетках (скелетные мышцы, миокард). Симптомы заболевания довольно вариабельны по времени своего проявления и выраженности у разных больных, традиционно наблюдается прогрессирующая мышечная слабость, при некоторых формах – кардиомегалия с дилятационной кардиомиопатией. Диагностика болезни Помпе производится на основании данных наследственного анамнеза, гистологического и гистохимического изучения мышечных тканей, биохимического анализа крови, а также генетических исследований. Лечение в настоящий момент может производиться с помощью фермент-заместительной терапии, однако эффективность этой методики неодинакова у разных пациентов.
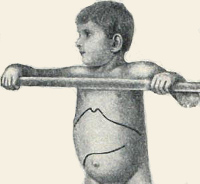
Общие сведения
Болезнь Помпе (гликогеноз 2-го типа, недостаточность кислой альфа-глюкозидазы) – наследственное заболевание, при котором из-за нарушения процессов обмена гликогена происходит повреждение нервных и мышечных тканей. Впервые было описано в 1932 году голландским ученым И. Помпе, с тех пор официально зарегистрировано более 50 случаев патологии. Болезнь Помпе с равной степенью вероятности поражает как мужчин, так и женщин, встречаемость колеблется от 1:60000 (взрослая форма) до 1:140000 (ранняя, или инфантильная форма). Является одной из немногих лизосомных болезней накопления, в отношении которой было разработано эффективное специфическое лечение, одобренное в США в 2006 году и в России – в 2013 г. Однако стоимость этиотропной терапии болезни Помпе крайне высока и составляет несколько сотен тысяч долларов в год. Смертность в случае отсутствия лечения зависит от формы патологии – детская инфантильная форма часто приводит к летальному исходу на 1-2-м году жизни ребенка, при типе болезни с отсроченным началом нарастание симптомов идет намного медленнее.
Причины болезни Помпе
Болезнь Помпе является классическим гликогенозом, при ней в тканях скелетных мышц, миокарда и отчасти нервной системы формируются отложения гликогена по причине невозможности его расщепления. Это происходит в результате мутации гена GAA, расположенного на 17-й хромосоме – он кодирует последовательность кислой альфа-1,4-глюкозидазы или мальтазы. Это один из ключевых ферментов лизосом, участвующий в расщеплении молекулы гликогена на более простые отрезки, которые, в конечном итоге, деградируют до глюкозы, вступающей в энергетический обмен клетки. Так как гликоген является важным депо энергии для таких структур, как скелетные мышцы, миокард, печень и нервная ткань, проявления болезни Помпе сводятся именно к патологическим изменениям данных органов.
В результате подобных изменений сначала возникает дефицит энергии в клетках – потребности тканей в глюкозе покрываются только за счет ее поступления из крови. Кроме того, в лизосомах при болезни Помпе начинает накапливаться гликоген, формируя крупные включения в виде вакуолей, в дальнейшем приводя к дистрофии и повреждению клеток. Наследование дефектных вариантов гена GAA происходит по аутосомно-рецессивному типу. Наличие нескольких форм заболевания предположительно объясняется разными типами мутаций вышеуказанного гена. Возможно, при некоторых дефектах происходит не полное исчезновение, а лишь снижение активности кислой альфа-1,4-глюкозидазы, что и приводит к более позднему развитию болезни Помпе и медленному прогрессированию заболевания. Определение формы патологии играет важную роль для составления ее прогноза и схемы лечения.
Классификация болезни Помпе
На сегодняшний день специалисты выделяют несколько основных форм болезни Помпе, основное различие между которыми заключается в сроках начала заболевания и выраженности симптомов. В большинстве случаев, с гликогенозом 2-го типа сталкиваются врачи-педиатры, однако имеется тип заболевания, выявляемый у взрослых.
Методами современной генетики на сегодняшний момент не определена взаимосвязь между отдельными типами мутаций гена GAA и формами болезни Помпе. Возможно, причина такой вариабельности проявлений лежит совсем в другом – в литературе описаны семейные случаи заболевания, когда у родственников регистрировались различные формы патологии. Изучение закономерностей, приводящих к развитию болезни Помпе определенного типа, сильно осложняется относительной редкостью данного синдрома.
Симптомы болезни Помпе
Проявления болезни Помпе довольно сильно отличаются при различных формах заболевания. Ранний инфантильный тип характеризуется выраженной мышечной слабостью младенца, снижением его двигательной активности, плаксивостью. В педиатрии при осмотре такого больного часто выявляется задержка психомоторного развития, различная степень увеличения печени, пальпация иногда выявляет гипертрофию мышц, которые, однако, при этом довольно слабые. При дальнейшем развитии болезни Помпе возникают проблемы с кормлением из-за слабости сосательной мускулатуры, выявляется дисфагия и, как итог всего этого – гипотрофия. Нарастающая кардиомиопатия и слабость дыхательной мускулатуры со временем приводят к смерти ребенка.
Поздняя инфантильная и ювенильная формы болезни Помпе протекают практически одинаково, различается только срок появления симптомов патологии. Как правило, выявляется мышечная слабость, признаки кардиомиопатии. Со временем начинает формироваться выраженная дистрофия скелетной мускулатуры, кардиомегалия, на этом фоне начинают увеличиваться печень и селезенка. Длительность течения этих форм болезни Помпе составляет около 10-12 лет, после чего, при отсутствии лечения, наступает летальный исход из-за декомпенсированной сердечной недостаточности. Косвенным симптомом будет являться большая частота простудных заболеваний с легочными осложнениями, ночное апноэ, головные боли по утрам.
Взрослая форма болезни Помпе характеризуется поздним началом (первые проявления возникают в 20-40 лет). Клинически раньше всего обнаруживается дистальная миопатия, проявляющаяся слабостью мышц конечностей. Со временем развиваются искривления позвоночника (сколиоз, лордоз), обусловленные слабостью окружающего позвоночный столб мышечного футляра. В некоторых случаях регистрируются медленно нарастающие признаки сердечной недостаточности, но при болезни Помпе такого типа поражение сердца возникает не всегда. Течение взрослой формы патологии занимает несколько десятилетий, поэтому в ряде случаев больные доживают до старости при приемлемом качестве жизни и без специфического лечения.
Диагностика болезни Помпе
Выявление болезни Помпе можно производить многочисленными клиническими методиками – биохимическим анализом крови, изучением биоптата мышц, культур фибробластов или лейкоцитов больного, традиционными исследованиями (осмотр, ЭКГ, ЭхоКГ). При осмотре часто обнаруживается слабость и дистрофия мышц, при вовлечении в патологический процесс внутренних органов – гепато- и спленомегалия. На электрокардиограмме регистрируется укорочение интервала PQ, расширение комплекса QRS, обусловленного увеличением размеров миокарда. По этой же причине увеличивается длительность фазы реполяризации желудочков, что проявляется инверсией зубца T. Эхографическое исследование сердца показывает резкое увеличение его размеров за счет значительного утолщения стенок желудочков. При взрослой форме болезни Помпе вышеуказанные изменения миокарда могут не выявляться.
Биохимический анализ крови позволяет обнаружить как специфические, так и косвенные признаки заболевания. Специфическим исследованием будет определение активности кислой альфа-1,4-глюкозидазы в плазме крови, которая при болезни Помпе будет резко снижена. Косвенным указанием на наличие патологии является резкое повышение активности креатинфосфокиназы, обусловленное поражением мышечной ткани. Гистологическое исследование биоптата мышц выявляет в миоцитах многочисленные включения гликогена, часто придающие им вид «пенистых клеток». Гистохимическое изучение при болезни Помпе выявляет резкое снижение активности кислой альфа-1,4-глюкозидазы в мышечной ткани, фибробластах и лейкоцитах.
Врачом-генетиком может быть проведено генетическое определение болезни Помпе – оно производится методом прямого секвенирования последовательности гена GAA с целью выявления дефектных участков. Кроме того, может помочь в диагностике заболевания и составление наследственного анамнеза. Генетическая диагностика болезни Помпе включает в себя секвенирование GAA и у фенотипически здоровых родственников больного с целью выявления носительства патологического гена.
Лечение и прогноз болезни Помпе
На сегодняшний день единственным методом специфического лечения болезни Помпе является фермент-заместительная терапия с целью восполнения дефицита кислой альфа-1,4-глюкозидазы. Для этого используют препарат альфа алглюкозидазы производства США. Стоимость этого лечения крайне высока (годовой курс стоит 100-400 тысяч долларов), однако его эффективность неодинакова у разных больных. Других способов лечения болезни Помпе в настоящее время не существует. Без лечения при инфантильных и ювенильной формах заболевания прогноз неблагоприятный, у взрослого типа – неопределенный. Профилактика возможна только путем своевременного выявления носительства болезни Помпе (в случае наличия патологии у кровных родственников) и последующей генетической пренатальной диагностики.
Глава 16. Болезнь Помпе: как не пропустить диагноз?
Несмотря на редкость патологии, вероятность встречи врача-педиатра и пациента с болезнью Помпе не так мала. Мизерное количество детей с установленным диагнозом (в Москве в настоящее время наблюдается только один ребенок с болезнью Помпе), вероятнее всего, связано с тем, что врачи не ждут ничего подобного и не замечают некоторых признаков. При этом вовремя установленный диагноз может спасти жизнь ребенку. Чтобы не пропустить пациента с болезнью Помпе, достаточно следовать приведенным ниже несложным правилам, сформулированным на основании анализа многих описанных случаев.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Болезнь Помпе (синонимы: недостаточность кислой мальтазы, гликогеноз II типа) — редкое, прогрессирующее наследственное заболевание, поражающее в первую очередь мышечную ткань. Код по МКБ10: Е74.0.
В основе патологического процесса лежит недостаточность фермента кислой альфаглюкозидазы (GAA) [1], расщепляющего гликоген в лизосомах клеток, в результате мутации в гене GAA, который кодирует данный фермент. Ген GAA локализован на длинном плече 17 хромосомы (17q25.2-q25.3), состоит из 20 экзонов и имеет размер 20 Кб. На сегодняшний день идентифицировано более 300 мутаций гена.
Частота болезни Помпе существенно зависит от этнической принадлежности и географического расположения и колеблется от 1 на 14 000 до 1 на 300 000 живых новорожденных [2, 8, 9]. Частота инфантильной формы выше среди афроамериканцев [2], в то время как болезнь Помпе с поздним дебютом чаще встречается в Нидерландах [8]. В среднем частота всех форм болезни Помпе оценивается как 1 на 40 000 живых новорожденных. Заболевание имеет аутосомно-рецессивный тип наследования. Это единственное заболевание из группы гликогенозов, которое относится к лизосомным болезням накопления. В связи с выраженной мышечной слабостью, гипотонией, болезнь Помпе также была отнесена к нервно-мышечным заболеваниям или метаболическим миопатиям.
При болезни Помпе гликоген накапливается в лизосомах поперечно-полосатой мускулатуры, сердечной мышцы, гладкой мускулатуры [2, 3].
Тяжесть заболевания во многом зависит от возраста появления первых симптомов, степени вовлеченности в патологический процесс органов и тканей и скорости прогрессирования.
КЛАССИФИКАЦИЯ И КЛИНИЧЕСКАЯ КАРТИНА
Разнообразие клинических проявлений болезни Помпе привело к формированию разделения заболевания на два основных подтипа, в зависимости от возраста дебюта и степени вовлечения в патологический процесс различных органов и тканей: инфантильная форма и форма болезни с поздним началом.
Классическая инфантильная форма — именно она была впервые описана профессором Помпе (P.J. Pompe), немецким врачом, в 1932 году [4] как быстро прогрессирующее заболевание, характеризующееся кардиомегалией, гепатомегалией, мышечной слабостью, гипотонией и смертью от сердечно-легочной недостаточности на первом году жизни [5, 6]. Неклассический вариант инфантильной формы — более доброкачественная, медленно прогрессирующая форма заболевания, характеризующаяся менее выраженной кардиомиопатией по сравнению с классической инфантильной формой, также проявляющаяся на первом году жизни. Эта форма была впервые описана Hers, а затем более детально Slonim с соавторами [7].
Болезнь Помпе с поздним дебютом
Детская, ювенильная форма или мышечный вариант — гетерогенная группа, характеризуется началом симптомов после первого года жизни и, как правило, отсутствием тяжелой кардиомиопатии.
Взрослая форма характеризуется медленно прогрессирующей миопатией с преимущественным поражением скелетных мышц, дебютирует в возрасте 2.
Болезнь Помпе
Это редкое генетическое заболевание, относящееся к группе лизосомных болезней. Характерно повсеместное поражение нервных и мышечных клеток из-за накопления гликогена. Симптомы болезни Помпе вариативны, но в целом характерна прогрессирующая мышечная слабость, а для некоторых форм заболевания кардиомегалия и дилатационная кардиомиопатия. Лечение болезни Помпе осуществляется фермент-заместительной терапией, однако эффективность отличается у разных пациентов.
Причины болезни Помпе
Болезнь Помпе — это классический гликогеноз — заболевание, при котором нарушено расщепление гликогена. Как следствие, происходит его накопление и отложение в организме. Патология развивается в результате мутации гена GAA, располагающегося на 17-й хромосоме и отвечающего за кодирование фермента мальтазы.
Мальтаза представляет собой ключевой лизосомный фермент, который расщепляет гликоген до более простых отрезков. Они метаболизируются до глюкозы и участвуют в энергетическом обмене клетки. При болезни Помпе сначала развивается дефицит глюкозы в клетках, а потом там начинает накапливаться гликоген, что приводит к развитию клеточной дистрофии.
Классификация болезни Помпе
Клиническая классификация болезни Помпе основана на возрасте манифестации проявлений. Различают следующие формы:
Симптомы болезни Помпе
Симптомы болезни Помпе различаются в зависимости от формы патологии. Для ранней инфантильной характерна выраженная слабость мышц. Ребенок становится плаксивым, гиподинамичным. Педиатры часто диагностируют задержку психомоторного развития. По мере прогрессирования заболевания из-за слабости сосательной мускулатуры присоединяются проблемы с кормлением, как следствие, развивается общая гипотрофия. Со временем нарастает кардиомиопатия и слабость дыхательной мускулатуры.
Что касается поздней инфантильной и ювенальной форм болезни, то они протекают со сходной симптоматикой и отличаются только возрастом начала клинической манифестации. Обычно диагностируется мышечная слабость и признаки миопатии сердца. По мере прогрессирования этих симптомов увеличивается печень и селезенка.
Взрослая форма болезни Помпе имеет наиболее благоприятный прогноз в плане продолжительности жизни. Из симптомов раньше всего обнаруживают дистальную миопатию, которая проявляется слабостью мышц конечностей. Постепенно в процесс вовлекаются мышцы туловища, в частности спины, что приводит к развитию сколиоза. Сердце и печень поражаются не всегда.
Диагностика и лечение болезни Помпе
Диагностика болезни Помпе основывается на клинических симптомах, лабораторном тестировании и инструментальном обследовании (ЭКГ). Специфичным исследованием является определение кислой альфа-1,4-глюкозидазы в сыворотке крови и гликогена в биоптатах мышечной ткани.
Из генетических исследований выполняется секвенирование гена GAA для выявления в нем мутаций. Данное тестирование назначается не только больным пациентам, но и их клинически здоровым родственникам для определения носительства мутации. Выполнить этот анализ можно в медико-генетическом центре «Геномед».
Единственный метод лечения на сегодня — это фермент-заместительная терапия, которая восполняет недостаточность кислой альфа-1,4-глюкозидазы. Терапия дорогостоящая. Что касается прогноза, то он зависит от формы болезни. Ранняя инфантильная наиболее неблагоприятна. При отсутствии лечения такие пациенты редко доживают до двух лет.
Наиболее благоприятно протекает взрослая форма заболевания. Она прогрессирует медленно, поэтому такие пациенты доживают до старости без специфического лечения и с сохранением приемлемого качества жизни.
Болезнь Помпе
Общая информация
Краткое описание
Российское общество медицинских генетиков
Год утверждения (частота пересмотра): 2017 (пересмотр каждые 3 года)
Болезнь Помпе (БП), также широко известная как гликогеноз II типа, относится к редким мультисистемным наследственным болезням накопления, связанным с дефицитом фермента кислой мальтазы (кислой альфа-глюкозидазы) в лизосомах. Преимущественное накопление гликогена отмечено в скелетных мышцах, но в разной степени может обнаруживаться и в других органах и тканях, включая сердечную мышцу, печень, нервную систему, гладкую мускулатуру и т.п.

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Этиология и патогенез
Мутации гена GAA приводят к снижению или полному отсутствию активности КАГ. Заболевание моногенное, наследуется по аутосомно-рецессивному типу.
Эпидемиология
Диагностика
• прогрессирующая мышечная слабость (особенно мышцы поясов конечностей и проксимальные отделы мышц конечностей).
| Клинический симптом | Частота |
| Синдром «вялого ребенка», быстрое прогрессирование мышечной слабости, аксиальная гипотония, снижение моторной активности, слабость мимических мышц, арефлексия в поздней стадии заболевания, икроножные мышцы плотные при пальпации | до 96% |
| Кардиомегалия (перкуссия) и признаки сердечной недостаточности | до 95% |
| Гепатомегалия (перкуссия, пальпация) | до 82% |
| Макроглоссия | до 62% |
| Трудности при вскармливании и низкая прибавка в весе | до 50% |
| Частые респираторные инфекции, респираторный дистресс и снижение дыхательной функции | 30-40% |
| Быстрое и неуклонное прогрессирование заболевания | 100% |
Достаточно часто к описанным выше изменениям присоединяются неспецифические нарушения в виде общего недомогания, раздражительности ребенка, повышенной потливости, рвоты и запоров. Редким, но настораживающим признаком полиорганной патологии является снижение слуха. В результате нарушения резорбции спинномозговой жидкости развивается гидроцефалия.
Особенности физикального обследования при БППН
| Предъявляемое задание | Выполняет без труда | Выполняет с трудом | Не может выполнить |
| Поднять руки над головой | 55% | 29% | 16% |
| Принять вертикальное положение из положения наклонившись вперед | 14% | 45% | 41% |
| Встать с низкого стула | 12% | 53% | 35% |
| Встать без помощи рук из положения лежа на спине | 8% | 37% | 55% |
| Подпрыгнуть на месте | 6% | 29% | 65% |
| Ходьба вверх/вниз по лестнице | 2% | 57% | 41% |
| Поднять ноги в положении лежа | 2% | 43% | 55% |
| Подняться с корточек | 2% | 22% | 76% |
| Признак | Частота |
| Прогрессирующая мышечная слабость с преимущественным поражением проксимальных отделов, снижение моторной активности, слабость в ногах больше, чем в руках, гипертрофия икроножных мышц, вовлечение параспинальных мышц (у детей постарше), гипотония, снижение сухожильных рефлексов, положительные приемы Говерса, миопатическая походка | 30-90% |
| Нарушения дыхания, частые респираторные инфекции, дыхательная недостаточность, диспноэ при физической нагрузке, обструктивное апноэ во время сна, ортопноэ | 25-40% |
| Умеренная гепатомегалия (пальпация, перкуссия) | до 16% |
| Кардиомегалия (перкуссия) | 0-12% |
| Макроглоссия | до 4% |
| Сонливость, утомляемость | до 8% |
| Лордоз, кифоз и/или сколиоз | 9-25% |
| Нормальное психоречевое развитие, сохранный интеллект | 96% |
Как уже говорилось выше, параспинальные мышцы и мышцы передней стенки живота могут поражаться на самых ранних стадиях БППН, что приводит к быстрой утомляемости, снижению переносимости стандартных нагрузок как на ранних, так и развернутых стадиях болезни. Важным является обнаружение нарушения дыхательной функции вследствие слабости мышц диафрагмы и межреберных мышц. До 1/3 взрослых пациентов с БППН имеют нарушения со стороны дыхательной системы.
Дифференциальный диагноз
Лечение
Комментарий: Снижение минеральной плотности костей является частой находкой у пациентов с болезнью Помпе и по данным последних исследований достигает уровня 67% случаев[2]. Остеопения и остеопороз обнаруживаются как при МБП, так и у детей и взрослых с БППН. Неустойчивость при ходьбе, сложность поддержания равновесия в результате мышечной слабости, в сочетании с остеопорозом угрожает развитием переломов костей и позвоночника. Обязательным является своевременное назначение препаратов витамина D, кальция, бисфосфонатов по схемам, рекомендованным для общей популяции.
Уровень убедительности рекомендации – 1 (уровень достоверности доказательств С).