Болезнь Дарье
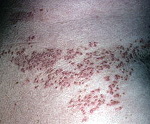
Болезнь Дарье (фолликулярный вегетирующий дискератоз) – наследственное заболевание, передающееся по аутосомно-доминантному типу с различной степенью фенотипического проявления дефектного гена. Основу клинических проявлений составляет патологическое ороговение клеток эпидермиса (дискератоз). Первичным элементом являются небольшие шелушащиеся пятна или шаровидные папулы, покрытые корками, сливающиеся в бляшки. Локализуется дерматоз на лице, груди, спине, в межлопаточной области, на волосистой части кожи головы. Возможно присоединение вторичной инфекции. Гендерного деления нет. Лечение симптоматическое. Прогноз для жизни благоприятный.
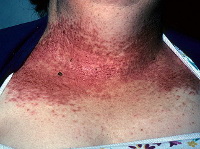
Общие сведения
Болезнь Дарье – редкий наследственный дерматоз, обусловленный аномальной мутацией части ДНК, отвечающей за синтез белка, связывающего клетки эпидермиса, с исходом в дискератоз. Прослеживается это наследственное заболевание в нескольких поколениях семьи, проявляется на двадцатом году жизни. Впервые патологию описал в 1889 году французский врач-дерматолог Ж. Дарье. Это был трактат о псороспермии, особых микроорганизмах – комочках протоплазмы, которую исследователь расценил как первопричину заболевания. Современные врачи-дерматологи хорошо знают, что болезнь Дарье – это генетическое заболевание, возникающее из-за дефекта в наследственном аппарате клетки, поражающее и женщин, и мужчин. Актуальность заболевания связана с наследственным характером процесса, требующим внимательного и ответственного отношения родителей к своим детям, уже растущим рядом с ними или ещё только планируемым к рождению. Сведений о распространённости заболевания нет.
Причины болезни Дарье
Генетическая предрасположенность, безусловно, является фактором риска в развитии болезни Дарье. Однако суть наследственного дерматоза заключается в нарушении межклеточных коммуникаций (десмосом). В результате мутации «белкового» сегмента ДНК (12g23-g24) нарушается функция кальциевых насосов (мембранных белков клетки), обеспечивающих прочность прилегания клеток друг к другу. Меняется структура эпидермиса: эпидермальные клетки размножаются, не успевая созреть, возникает дискератоз. Некоторые дерматологи склонны считать дополнительной причиной развития дискератоза гиповитаминоз А. Потерянная плотность эпидермиса ведёт к образованию межклеточных промежутков, куда немедленно мигрирует патогенная флора, инфицируя здоровую кожу. Кроме того, постоянно вегетирующие клетки рогового слоя начинают самоотслаиваться, обнажая эрозивную поверхность, которую также инфицируют микробы.
Классификация болезни Дарье
В зависимости от клинических проявлений наследственный дерматоз делят на:
Симптомы болезни Дарье
Первым проявлением фолликулярного вегетирующего дискератоза считают расширение устья потовых желёз – появление хорошо заметных углублений на неизменённой внешне коже. Дебютом болезни Дарье называют и поражение ногтей с отчётливым чередованием розоватых и белесых продольных полос (лейконихия) на поверхности ногтя, канавками, трещинами, подногтевым гиперкератозом. Иногда клиновидный дефект в виде щели буквально «режет» ноготь пополам.
Классическая форма заболевания проявляется высыпанием фолликулярных папул, покрытых струпьями (корками) на участках кожного покрова, типичных для локализации себорейного процесса: лоб, носогубные складки, виски, шея, область декольте, межлопаточное пространство. Поражается также волосистая часть кожи головы. При снятии корок образуются небольшие эрозии, которые воспаляются из-за присоединения вторичной инфекции, становясь причиной боли, неприятного запаха, общей интоксикации организма.
При локализованной или абортивной форме заболевания высыпания располагаются линейно только на ограниченном участке кожи: первичный элемент папула, вторичный – пигментация и эрозия. Изолированная форма характеризуется сочетанием узелков и полигональных папул, напоминающих бородавки и имеющих тенденцию к слиянию в бляшки. Локализуется процесс на тыле кистей и стоп. При этом ладони и подошвы поражаются точечным кератозом диффузного характера.
При везикулёзно-буллёзной разновидности болезни Дарье к узелкам присоединяются пузырьки с прозрачным содержимым. Сыпь локализуется в крупных складках кожи и на слизистых. Если процесс поражает исключительно слизистые оболочки, говорят о лейкоплакии, требующей к себе особого внимания.
В качестве фоновой патологии фолликулярного вегетирующего дискератоза встречается катаракта, миопия, нистагм, косоглазие, заболевания эндокринных желёз, эпилептические припадки, деменция, кисты костной ткани. Дерматоз имеет волнообразное течение с длительными ремиссиями и обострениями. Провоцирующим моментом чаще всего является УФО.
Диагностика и лечение болезни Дарье
Диагноз заболевания ставят на основании анамнеза, типичных клинических проявлений патологии с гистологическим подтверждением (при необходимости). Дифференцируют с красным плоским лишаём, «сухой» и грибковой экземой, семейной доброкачественной пузырчаткой Гужеро-Хейли-Хейли, болезнью Гровера, акрокератозом Гопфа.
Лечение симптоматическое, длительное и комплексное. В первую очередь, необходимо исключить факторы, способные спровоцировать обострение или рецидив заболевания: УФО, переохлаждение, высокая влажность, потливость. Далее – избавиться от вторичной инфекции, если таковая есть, с помощью антибиотиков ( амоксициллин+клавулановая кислота), противогрибковых препаратов (кетоконазол). Местно используют анилиновые красители, аэрозоли на основе антибиотиков и гормонов. При выраженном воспалительном процессе с экссудативным компонентом применяют глюкокортикоиды внутрь.
Параллельно санируют очаги хронической инфекции и лечат сопутствующую соматическую патологию. Основное внимание уделяют коррекции ороговения, применяя витамин А в больших дозах, витамин Е. Наружно назначают салициловые мази (2%-5%), крем Унны с ретинолом, диметилсульфоксид. Показана оксигенотерапия, кислородные и морские ванны. Рекомендована ПУВА-терапия. Элементы больших размеров удаляют после консультации с врачом-дерматологом и дерматокосметологом. Прогноз благоприятный. Под влиянием терапии или самопроизвольно в некоторых случаях возможны длительные и стойкие ремиссии, однако выздоровление невозможно.
Болезнь дискератоз что это
Дискератоз фолликулярный, известный также как болезнь Дарье (БД), относится к наследственным заболеваниям и характеризуется нарушением процессов ороговения в эпидермисе, клинически проявляющимся явлениями дискератоза. Впервые дерматоз был описан в 1889 г. J. Darier во Франции и J. White в США, которые не только описали, но и доказали генетическую природу дискератоза. Как правило, заболевание начинается в детском или в юношеском возрасте от 6 до 20 лет, однако описаны случаи возникновения и в 4 года, и в возрасте 70 лет. Примечательно, что первый случай врожденного фолликулярного дискератоза был диагностирован при биопсии у ребенка с отягощенным семейным анамнезом, когда БД страдали родственники больного по крайней мере в трех поколениях [1]. Заболевание не имеет гендерных отличий и встречается во всем мире. Распространенность по различным оценкам составляет 1 случай на 30 000-100 000 населения [2].
Этиология и патогенез
В качестве основной причины болезни Дарье на сегодняшний день рассматривают мутацию гена ATP2A2 хромосомы 12q23-24.1, кодирующей саркоплазматический/эндоплазматический ретикул Ca 2+ АТФ 2-го типа (SERCA2), который является кальциевым насосом [3]. Несмотря на то что данная мутация в гене ATP2A2 была идентифицирована у достаточно большого количества пациентов, попытки генотипировать ее оказались безуспешными. Поскольку клинические проявления и степень тяжести дискератоза в значительной степени могут варьировать даже у членов одной семьи при наличии идентичной мутации, по-видимому, в механизмах возникновения играют роль как другие гены, так и факторы фенотипического характера [4, 5]. Исследования последних лет доказали роль генных белков Bcl-2, отвечающих за регулирование апоптоза эпидермальных кератиноцитов в развитии БД [6].
Первые поражения кожи, зачастую сопровождающиеся зудом, как правило, отмечаются в подростковом возрасте. Основными триггерными факторами являются теплый климат, влажность, повышенная потливость, инсоляция, воздействие ультрафиолетовых лучей В, прием препаратов лития, системных кортикостероидов, эстрогенов, механическая травма, психоневрологические нарушения [7, 8].
Клиническая картина
В зависимости от преобладания тех или иных элементов сыпи принято выделять следующие клинические формы:
— везикулярная (везикуло-буллезная, пемфигоидная);
— абортивная или линеарная.
Изменения ногтей при БД имеют важное диагностическое значение. Белые и красные продольные полосы, продольное расщепление и подногтевой гиперкератоз являются наиболее патогномоничными признаками. Ногтевые пластинки пальцев кистей и стоп нередко изменяются по типу онихогрифоза. Слизистые оболочки поражаются редко, примерно у 15% больных, и не сопровождаются какими-либо субъективными ощущениями. В таких случаях на слизистой оболочке полости рта, гортани, зева и даже пищевода возникают лентикулярные белесовато-синеватые папулы, светло-белые узелки или белые линейные утолщения, напоминающие лейкоплакию [9].
Гистопатология
При электронно-микроскопических исследованиях доказано, что в основе патологических изменений лежат специфические процессы разрушения тонофиламенто-десмосомных комплексов. Вследствие этого клетки теряют десмосомный аппарат и обусловливают развитие акантолиза с последующим нарушением кератинизации.
Дифференциальный диагноз
БД имеет выраженное клиническое и гистологическое сходство с доброкачественной семейной пузырчаткой Хейли-Хейли, но у пациентов с БД высыпания появляются в более раннем возрасте и помимо складок располагаются и на других участках тела. Многие дерматологи связывают эти два диагноза, считая буллезную форму БД своеобразной переходной стадией к пузырчатке Гужеро-Хейли-Хейли. При наличии буллезных высыпаний необходимо помнить, что первичным элементом БД является папула, покрытая коркой-чешуйкой. Дополнительным критерием в пользу БД может послужить «волосатый язык», высыпания в полости рта по типу лейкоплакии, симптом Бенье. В особых случаях необходима биопсия кожи с последующей гистологией.
Акрокератоз бородавчатый Гопфа относится к редким генодерматозам с аутосомно-доминантным типом наследования. Заболевание чаще начинается в детском возрасте с высыпаний на тыле кистей и стоп, в дальнейшем высыпания распространяются на кожу задней поверхности предплечий, ладоней и подошв, коленей, голеней. Первичным элементом являются папулы полигональной формы с гладкой или бородавчатой поверхностью, бурого или красновато-коричневого оттенка, сливающиеся в бляшки. Возможно линейное расположение высыпаний, точечный кератоз. Также как и при БД возможно изменение ногтевых пластин. Сходность клинических картин этих двух дерматозов дает основание многим авторам считать веррукозный акрокератоз Гопфа вариантом БД. В патоморфологической картине характерно образование супрабазальных щелей, дискератоз, акантоз, папилломатоз, гранулез.
Верруциформная эпидермодисплазия Левандовского–Лютца также в большинстве случаев начинается в детском возрасте и характеризуется появлением множественных бородавчатых элементов мягкой консистенции на коже лица, шеи и дистальных поверхностей конечностей, цвета нормальной кожи, розового или светло-коричневого. Узелки могут сливаться в бляшки различной величины и формы. Кроме бородавчатых высыпаний на коже туловища и лица встречаются пигментные пятна по типу лентиго, возможно возникновение алопеции. Правильно поставить диагноз помогает отличная от БД локализация высыпаний и отсутствие плотных корко-чешуек на поверхности.
Терапевтические методики
На сегодняшний день терапевтические мероприятия при БД включают наружное лечение, системную терапию, фототерапию, профилактику и лечение вторичного инфицирования очагов.
Актуальными наружными средствами являются кортикостероиды, топические ретиноиды (адапален, 0,01% гель тазаротен, третиноин), в том числе в комбинации с кортикостероидами 13. Эти препараты регулируют процессы регенерации, уменьшают деление гиперпролиферирующих кератиноцитов и модулируют их дифференцировку.
При присоединении вторичной инфекции показаны комбинированные с антибиотиками и антимикотиками средства. Имеются сведения об эффективности применения 5-фторурацила при ограниченных формах БД [15, 16] и лосьона кальципотриола [17]. В последние годы в качестве альтернативы топическим кортикостероидам при стероид-чувствительных дерматозах активно используются ингибиторы кальциневрина. Так, при БД хорошую эффективность показал пимекролимус [18]. Одним из перспективных направлений в наружном лечении больных БД является применение селективных ингибиторов ЦОГ-2 (нестероидные противовоспалительные препараты), которые восстанавливают снижающуюся регуляцию экспрессии гена ATP2A2/SERCA2 в кератиноцитах [19]. Так, ученые из Испании [20] сообщили об успешном использовании ингибитора ЦОГ-2 диклофенака натрия (в виде 3% геля) у пациентов с БД.
Системными препаратами выбора при БД являются ретиноиды (ацитретин, изотретиноин, этретинат, алитретиноин) 21. Эти препараты являются самыми эффективным для лечения БД. У 90% пациентов достигается максимальное уменьшение симптомов. Препараты нормализуют терминальную дифференцировку клеток, тормозят пролиферацию эпителия протоков сальных желез, образование детрита, облегчают его эвакуацию, а также снижают выработку кожного сала и облегчают его выделение.
Длительное использование системных ретиноидов ограничено из-за их побочных и нежелательных действий, таких как сухость слизистых оболочек, светочувствительность, гиперлипидемия, нарушения функции печени, остеопороз. Системные ретиноиды обладают тератогенным действием и необходимостью назначения оральных контрацептивов после консультации гинеколога. В ряде случаев для усиления эффекта одновременно с ретиноидами назначают ПУВА-терапию, однако данный метод из-за выраженной иммуносупрессии применяется достаточно редко. При отдельных веррукозных очагах проводится хирургическое иссечение.
Прогноз
Болезнь протекает неопределенно долго (много лет) и обычно не вызывает заметных субъективных ощущений и нарушения общего состояния. Может наступить спонтанная полная клиническая ремиссия. Чаще всего болезнь отступает не в результате терапии, а с возрастом. Серьезным осложнением, связанным с БД, является повышенная восприимчивость к бактериальным и вирусным инфекциям, в частности к вирусу простого герпеса.
Врожденный дискератоз
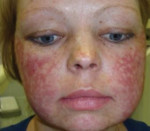
Врожденный дискератоз – крайне редкое генетическое заболевание, классическая клиника которого включает локальную гиперпигментацию кожных покровов, дистрофию ногтевых пластинок и лейкоплакию. Также возникает депрессия костного мозга, что проявляется апластической анемией, иммунодефицитом и кровотечениями. Часто врожденный дискератоз осложняется злокачественными новообразованиями. Диагностика подразумевает сопоставление клинических симптомов и выявление дефектов кариотипа при помощи кариотипирования или флуоресцентной гибридизации. Лечение заключается в восстановлении нормальной функции костного мозга путем его трансплантации или фармакологической стимуляции. Параллельно проводится симптоматическая терапия.
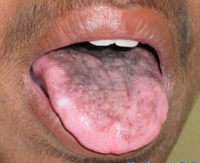
Общие сведения
Врожденный дискератоз – генетически обусловленный синдром, проявляющийся недостаточностью костного мозга и совокупностью специфических симптомов. Впервые классическая триада заболевания была описана в 1906 году немецким дерматовенерологом Цинссером. В 1930-х годах Коул и Энгман дополнили описание, после чего патология получила второе название – синдром Цинссера-Коула-Энгмана. На данный момент клиническая характеристика заболевания, помимо классической триады, включает другие симптомы и синдромы. Патология встречается крайне редко, распространенность – 1:1000000. Представители мужского пола болеют в 4,7 раз чаще, чем женского. Манифестация заболевания происходит в возрасте от 5 до 15 лет, поэтому первым с ним сталкивается педиатр. Симптомы развиваются постепенно, наиболее выраженная клиническая картина приходится на возраст 25-30 лет и старше. Как правило, больные умирают от осложнений в виде вторичных инфекций, злокачественных новообразований и внутренних кровотечений.
Причины врожденного дискератоза
Симптомы врожденного дискератоза
Классический вариант врожденного дискератоза включает в себя триаду симптомов: дистрофические изменения ногтей, гиперпигментацию кожных покровов и лейкоплакию слизистых оболочек. Более чем в 85% случаев его течение осложняется другими патологическими состояниями: апластической анемией, иммунодефицитом, аномалией развития мозжечка, умственной отсталостью, фиброзным перерождением легких или печени, злокачественным новообразованием. Заболевание развивается постепенно. До 5 лет врожденный дискератоз протекает латентно. Первые проявления могут развиваться в возрасте от 6 до 15 лет, иногда – после 20. С возрастом происходит усугубление всех симптомов и развитие осложнений.
Первичным проявлением врожденного дискератоза, как правило, выступает дистрофия ногтевых пластинок. Ногти становятся тонкими и ломкими, возникает продольная исчерченность. Спустя 2-3 года к данным проявлениям присоединяется сетчатая гиперпигментация кожи с характерной локализацией – лицо, шея, верхняя часть поверхности грудной клетки. Средний размер пораженных участков – 2-8 мм в диаметре. Кожа атрофируется, приобретает коричневый оттенок, на ней могут наблюдаться телеангиэктазии. Часто возникает гипергидроз и гиперкератоз стоп и ладоней, акроцианоз, алопеция. Спустя некоторое время ногтевые пластики могут самопроизвольно отпадать, что чаще всего наблюдается на мизинцах стоп.
Более чем у половины больных в возрасте до 8-10 лет развивается гипопластическая анемия, которая в педиатрии часто расценивается как анемия Фанкони. Первые клинические проявления – частые носовые кровотечения. Вскоре происходит снижение иммунитета. Изредка анемия и иммунодефицит являются первичными признаками врожденного дискератоза. В возрасте 25-30 лет возникает поражение слизистых оболочек рта. На них могут развиваться везикулы или папулы, переходящие в эрозии; папилломы, атрофия сосочков языка, гингивит, лейкоплакия. Наблюдается выпадение зубов. Часто возникает поражение глаз – конъюнктивит, блефарит, эктропион, катаракта и глаукома. Постепенно лейкоплакия поражает и другие слизистые оболочки – мочеиспускательного канала, кишечника, половых органов. У трети больных наблюдаются умственная отсталость.
Диагностика врожденного дискератоза
Диагностика врожденного дискератоза затруднена. Заболевание встречается крайне редко и имеет большое количество клинических проявлений. Диагноз выставляется на основании всех имеющихся симптомов и синдромов, данных лабораторных анализов и гистологического исследования биоптата кожных покровов.
Общие клинические анализы позволяют установить наличие отдельных синдромов. В ОАК на ранних стадиях возможен лейкоцитоз, нейтропения, повышение СОЭ. Далее развивается панцитопения, апластическая анемия, возможны макроцитоз и повышение концентрации фетального гемоглобина. Стернальная пункция на поздних стадиях выявляет депрессию костного мозга. Гистологическое исследование пораженных участков кожи позволяет обнаружить атрофию эпидермиса, слабовыраженный гиперкератоз, увеличение пигментации базального слоя, в дерме – увеличение количества меланофагов в сосочковом и сетчатом слоях, расширение сосудов микроциркуляторного русла. Возможно нарушение структуры коллагена, гомогенизация и фрагментация его волокон. Специфическая диагностика направлена на выявление дефектов кариотипа. Проводится кариотипирование или флуоресцентная гибридизация, при которых удается установить мутацию гена в участке Xq28, редко – в 3q14.
Лечение врожденного дискератоза
Лечение врожденного дискератоза малоэффективно. Основная цель – борьба с угнетением функции костного мозга. Метод выбора – трансплантация костного мозга от родного брата или сестры, не страдающих данным заболеванием. Даже на фоне пересадки у большей части больных наблюдается реакция «трансплантат против хозяина». При невозможности провести трансплантацию показано использование анаболических гормональных средств (оксиметолон), колониестимулирующих факторов, эритропоэтинов. Лечение злокачественных новообразований у таких больных проводится при помощи протонной терапии. При лейкоплакии используются ретиноиды и Р-каротин.
Прогноз при врожденном дискератозе неблагоприятный. Почти всегда заболевание имеет летальный исход. К смерти приводят злокачественные новообразования, присоединение вторичной инфекции (в основном – пневмонии), желудочно-кишечные или другие внутренние кровотечения. Чаще всего пациенты умирают от плоскоклеточного рака ротовой полости, гортаноглотки, половых органов, мочеиспускательного канала и ЖКТ, реже – аденокарциномы поджелудочной железы, лимфомы Ходжкина.
Публикации в СМИ
Дискератоз врождённый
Врождённый дискератоз — наследственное заболевание кожи с пойкилодермией, лейкоплакией слизистой оболочки рта, дистрофией ногтей, ладонно-подошвенным гипергидрозом, закупоркой или атрезией слёзно-носовых каналов; возможно сочетание с тромбоцитопенической пурпурой. Чаще наблюдают Х-сцепленную форму.
• Дискератоз врождённый Х-сцепленный (*305000, Xq28, ген DKC, À рецессив). Клинически: сетчатая гипер- и гипопигментация кожи, атрофия кожи, телеангиэктазии, дистрофия ногтей, постоянное слезотечение вследствие атрезии слёзных протоков, тромбоцитопения, анемия, панцитопения, атрофия яичек, озлокачествляющаяся лейкоплакия заднего прохода, слизистой оболочки рта и кожи, болезнь Ходжкена, аденокарцинома поджелудочной железы, нарушения иммунитета (инфицирование условно-патогенной микрофлорой, повышение уровней иммуноглобулинов), умственная отсталость, глухота, пренатальное и послеродовое замедление роста, кровотечения из язв слизистой оболочки, цирроз печени, некроз головок бедренных костей, внутричерепные кальцификаты. Лабораторно: повышенная частота хромосомных нарушений, индуцированных радиацией. Синоним: синдром Цинссера–Коула–Энгмена.
• Дискератоз врождённый типа Скоггинса (127550, Â ). Клинически: ретикулярная гиперпигментация кожи, отсутствие дерматоглифических узоров на пальцах, ладонный гиперкератоз, дистрофия ногтей, остеопороз, лейкокератоз слизистых оболочек, редкие волосы, аномалии зубного ряда, отсутствие слёзных точек, анемия, иммунодефицит. Лабораторно: меланин в кожных фагоцитах, эндоредупликация хромосом, увеличенная частота хромосомных разрывов.
• Дискератоз врождённый доброкачественный (*127600, Â ) — проявляется лейкоплакией на слизистых оболочках ротовой полости и конъюнктивах. Лабораторно: шиповатый слой эпидермиса содержит многочисленные эозинофильные клетки.
• Кератоз фолликулярный (кератоз волосяной). Участки ороговения локализуются в области устьев волосяных фолликулов; заболевание обычно не создаёт для пациента серьёзных неудобств и представляет чисто косметическую проблему. Этиология • Неизвестна • Заболевание часто передаётся по наследству (например, 308800, Xp22.2–p22.13, ген KFSD, À доминантное). Клиническая картина • Множественные мелкие заострённые роговые папулы появляются в основном на боковых частях рук, бёдер и ягодиц • Участки ороговения могут быть на лице (особенно у детей) • Клиническая картина усиливается в холодную погоду и улучшается летом. Лечение • Не является необходимым, обычно неэффективно • Гидрофильный вазелин с водой (в равных соотношениях), кольдкрем, мазь с салициловой кислотой на вазелине (3%) • Гель салициловой кислоты (6%), забуференные лосьоны с молочной кислотой часто оказываются эффективными.
• Порокератоз Мибелли (*175800, Â ) — наследственный кератоз с преимущественным поражением эпидермиса в зоне выводных протоков потовых желёз, характеризуется возникновением бляшек с атрофией в центре и роговым периферическим валиком. Синонимы • Гиперкератоз эксцентричный • Гиперэлеидоз • Гиперэлеидоз эксцентричный атрофический • Невус кератоатрофический.
МКБ-10. L90.8 Другие атрофические изменения кожи
Код вставки на сайт
Дискератоз врождённый
Врождённый дискератоз — наследственное заболевание кожи с пойкилодермией, лейкоплакией слизистой оболочки рта, дистрофией ногтей, ладонно-подошвенным гипергидрозом, закупоркой или атрезией слёзно-носовых каналов; возможно сочетание с тромбоцитопенической пурпурой. Чаще наблюдают Х-сцепленную форму.
• Дискератоз врождённый Х-сцепленный (*305000, Xq28, ген DKC, À рецессив). Клинически: сетчатая гипер- и гипопигментация кожи, атрофия кожи, телеангиэктазии, дистрофия ногтей, постоянное слезотечение вследствие атрезии слёзных протоков, тромбоцитопения, анемия, панцитопения, атрофия яичек, озлокачествляющаяся лейкоплакия заднего прохода, слизистой оболочки рта и кожи, болезнь Ходжкена, аденокарцинома поджелудочной железы, нарушения иммунитета (инфицирование условно-патогенной микрофлорой, повышение уровней иммуноглобулинов), умственная отсталость, глухота, пренатальное и послеродовое замедление роста, кровотечения из язв слизистой оболочки, цирроз печени, некроз головок бедренных костей, внутричерепные кальцификаты. Лабораторно: повышенная частота хромосомных нарушений, индуцированных радиацией. Синоним: синдром Цинссера–Коула–Энгмена.
• Дискератоз врождённый типа Скоггинса (127550, Â ). Клинически: ретикулярная гиперпигментация кожи, отсутствие дерматоглифических узоров на пальцах, ладонный гиперкератоз, дистрофия ногтей, остеопороз, лейкокератоз слизистых оболочек, редкие волосы, аномалии зубного ряда, отсутствие слёзных точек, анемия, иммунодефицит. Лабораторно: меланин в кожных фагоцитах, эндоредупликация хромосом, увеличенная частота хромосомных разрывов.
• Дискератоз врождённый доброкачественный (*127600, Â ) — проявляется лейкоплакией на слизистых оболочках ротовой полости и конъюнктивах. Лабораторно: шиповатый слой эпидермиса содержит многочисленные эозинофильные клетки.
• Кератоз фолликулярный (кератоз волосяной). Участки ороговения локализуются в области устьев волосяных фолликулов; заболевание обычно не создаёт для пациента серьёзных неудобств и представляет чисто косметическую проблему. Этиология • Неизвестна • Заболевание часто передаётся по наследству (например, 308800, Xp22.2–p22.13, ген KFSD, À доминантное). Клиническая картина • Множественные мелкие заострённые роговые папулы появляются в основном на боковых частях рук, бёдер и ягодиц • Участки ороговения могут быть на лице (особенно у детей) • Клиническая картина усиливается в холодную погоду и улучшается летом. Лечение • Не является необходимым, обычно неэффективно • Гидрофильный вазелин с водой (в равных соотношениях), кольдкрем, мазь с салициловой кислотой на вазелине (3%) • Гель салициловой кислоты (6%), забуференные лосьоны с молочной кислотой часто оказываются эффективными.
• Порокератоз Мибелли (*175800, Â ) — наследственный кератоз с преимущественным поражением эпидермиса в зоне выводных протоков потовых желёз, характеризуется возникновением бляшек с атрофией в центре и роговым периферическим валиком. Синонимы • Гиперкератоз эксцентричный • Гиперэлеидоз • Гиперэлеидоз эксцентричный атрофический • Невус кератоатрофический.
МКБ-10. L90.8 Другие атрофические изменения кожи