Болезнь Александера
Болезнь Александера – это довольно редкое заболевание нервной системы, обусловленное генной мутацией. Симптомы развиваются в результате нарушения питания нервных клеток и блока проведения нервных импульсов по миелиновым волокнам.
Впервые заболевание было описано патологоанатомом Александером в 1949 году. Болезнь характеризуется неуклонно прогрессирующим течением. Распространенность болезни Александера достоверно не изучена ввиду малой встречаемости этой патологии. Описано около 500 случаев заболевания в США; проведенное в Японии исследование выявило наличие 1 случая заболевания на 2 700 000 человек. Теоретически подсчитана вероятность возникновения болезни Александера в человеческой популяции – 1:3 000 000. Точно известно, что возникновение заболевания не зависит от расы, пола, места проживания на Земном шаре.
Причины
В 95% случаев болезнь Александера развивается в результате мутации в гене, расположенном на 17-й хромосоме. Обычно мутация возникает спонтанно, то есть родители являются совершенно здоровыми, их генотип не имеет подобных изменений. Скорее всего, изменение гена происходит в отцовской хромосоме во время сперматогенеза, и если такой «аномальный» сперматозоид оплодотворяет яйцеклетку, то тогда и развивается болезнь у ребенка.
Ген отвечает за продукцию глиального фибриллярного кислого белка GFAP. В случае мутации измененный белок GFAP накапливается во вспомогательных клетках нейронов (нейроглии), что препятствует обеспечению нейронов питательными веществами. Кроме того, при болезни Александера в самом измененном белке GFAP образуются узелковые образования, которые называют волокнами Розенталя. Последние мешают нормальному проведению нервных импульсов по миелиновым волокнам.
У 5% людей, у которых диагностирована болезнь Александера, подобный или иной генетический дефект не обнаруживается, то есть причина развития остается неизвестной.
Клиническая картина
Заболевание впервые проявляет себя у людей в разном возрасте. В зависимости от этого принято выделять несколько клинических форм:
Предполагается наличие так называемой неонатальной формы заболевания, когда ребенок рождается уже с проявлениями болезни. У таких детей обычно с первых дней жизни отмечается повышенное внутричерепное давление, аномально большой череп. Характерен судорожный синдром, выраженная задержка нервно-психического развития. К сожалению, продолжительность жизни таких деток не составляет и года. Некоторые ученые относят эту форму к инфантильной, но с очень ранним началом.
Инфантильная форма развивается в раннем детском возрасте, в среднем – по достижении 6 месяцев. У таких детей плохой аппетит, они часто срыгивают вплоть до рвоты. Отмечается патологически быстрое увеличение размеров головы, нарастание внутричерепного давления. Естественно, что это сказывается на темпах физического и нервно-психического развития. Дети плохо прибавляют в весе, поздно начинают держать голову (после 3-х месяцев), садиться и ползать. По мере роста и развития ребенка развивается мышечная слабость в конечностях (парезы) наряду с повышенным мышечным тонусом (спастичность), что проявляется ограничением объема и силы произвольных движений. На фоне парезов в конечностях появляются непроизвольные движения: выкручивающие, червеобразные движения в пальцах рук, повороты головой с фиксацией позы и тому подобное. Эти явления называются гиперкинезами, в частности, хореоатетозом. Возможны судорожные эпилептические припадки. Страдает интеллект: дети не узнают близких, их не радуют игрушки, они не овладевают навыками (например, не могут нанизать кольца на пирамидку в соответствующем возрасте). Также нарушается координация движений, наблюдаются подергивания глазных яблок (нистагм). Самостоятельная ходьба практически невозможна. Заболевание неуклонно прогрессирует и заканчивается смертью в течение 2-3 лет.
Ювенильная форма проявляет себя несколько позже, в возрасте от 4 до 14 лет, в среднем – около 9 лет. Хотя отдельные признаки заболевания могут появиться и раньше – в 2-3 года, но обычно их не связывают с болезнью Александера. Такие детки несколько отстают в нервно-психическом развитии, страдают от судорог. У них голова имеет больший размер по сравнению со сверстниками (но не настолько больший по сравнению с инфантильной формой). Несколько позже присоединяются нарушения речи (смазанность, нечеткость), поперхивание при приеме пищи, а затем и при глотании воды. Голос приобретает гнусавый оттенок. Движения языком затрудняются. Все эти изменения формируют бульбарные и псевдобульбарные расстройства, а возникают в результате поражения ствола мозга. По утрам больных беспокоят неукротимые рвоты. Так же, как и при инфантильной форме, появляется мышечная слабость в конечностях, которая постепенно нарастает.
Мышечный тонус увеличивается, мышцы становятся плотными и твердыми на ощупь, появляются патологические стопные признаки (симптом Бабинского и другие). Постепенно эти изменения охватывают все четыре конечности, что становится причиной расстройств передвижения и самообслуживания. Возможны нарушение равновесия, расстройства поведения. Обычно интеллектуальные расстройства выражены незначительно или вообще отсутствуют, хотя описаны случаи и резкого снижения мыслительных способностей. У больных с ювенильной формой периодически регистрируют рефлекторную остановку дыхания: апноэ. В конце концов, прогрессирующее поражение нервной системы заканчивается смертельным исходом, в среднем, через 10 лет от появления начальных клинических признаков заболевания.
Взрослая форма развивается в сроки от 20 до 70 лет. Клинические симптомы довольно разнообразны, поскольку могут быть отражением патологии любого участка головного мозга. Чаще всего это парезы и параличи с повышенным мышечным тонусом, нарушения координации движений и равновесия, непроизвольные неконтролируемые движения, нарушения речи и глотания. Снижение интеллекта незначительное. Часто выявляется нистагм и нарушение содружественных (одновременных и однонаправленных) движений глазными яблоками. Болезнь прогрессирует и неизбежно заканчивается летальным исходом (обычно от присоединения интекуррентных инфекций).
Диагностика
Диагностика заболевания прижизненно довольно затруднительна, потому что клинических симптомов, свойственных только болезни Александера, нет. И специфических изменений ни один из методов исследования не выявляет (не считая генетического анализа, который, тем не менее, необходимо еще назначить, подозревая эту болезнь).
При магнитно-резонансной томографии головного мозга (МРТ) при болезни Александера выявляется демиелинизация различных отделов мозга (при инфантильной и юношеской формах — преимущественно в лобных с распространением на другие области, при взрослой – более выражена в мозжечке и стволе мозга).
При электроэнцефалографии регистрируют изменения биоэлектрической активности мозга в лобных отделах.
Генетический анализ наиболее точно позволяет подтвердить диагноз болезни Александера: находят мутацию в гене GFAP на 17-й хромосоме (в 95% случаев). Следует помнить, что у 5% больных этим заболеванием генетический дефект не обнаружен и по сей день.
Подтверждением заболевания служит обнаружение волокон Розенталя (что возможно при биопсии мозга или уже после смерти при вскрытии).
Лечение
Сегодня медицина не располагает эффективными способами лечения болезни Александера. Возможно, будущее в этом направлении принадлежит генной инженерии.
После установления диагноза обычно проводят симптоматическую терапию, позволяющую облегчить и продлить жизнь больному:
Для передвижения используют специальные приспособления, в том числе и ортопедические. Пик болезни позволяет передвигаться только с помощью инвалидной коляски. Конечно, в терминальных стадиях заболевания больные нуждаются в постоянном постороннем уходе.
Как теперь будут лечить болезнь Александера
Ученые знают, что генетические мутации, приводящие к выработке дефектного белка под названием GFAP, вызывают болезнь Александера. Это тяжелое нейродегенеративное состояние – нервная система повреждается и перестает правильно работать. Патология может проявляться в младенчестве, подростковом или взрослом возрасте.

Многие люди с этим редким заболеванием умирают в течение первых нескольких лет, но некоторые живут с ним в течение нескольких десятилетий. В настоящее время исследователи Медицинской школы UNC изучают различия в обменных процессах пациентов с тяжелыми и легкими формами болезни. Международная группа ученых обнаружила, что мутантная форма белка GFAP имеет различные химические модификации в зависимости от времени появления симптомов.
Впервые ученые смогли смоделировать очень специфические химические изменения в GFAP, которые происходят внутри мозга больных людей. Это позволяет исследовать детали того, как неправильное образование и накопление GFAP изменяет работу клеток, приводя к прогрессированию заболевания и смерти. Сейчас они продолжают исследовать ферменты, ответственные за ключевые реакции в клетках мозга, которые приводят к болезни Александера. Результаты могут открыть двери для разработки лекарств и, в конечном итоге, для новых видов терапии пациентов с этим ужасным заболеванием.
Что такое болезнь Александера
Это лейкодистрофия, редкая группа расстройств нервной системы, которые включают разрушение миелина, жировой оболочки, изолирующей длинные отростки нервных клеток. Именно они осуществляют быструю и точную передачу электрических импульсов по всей нервной системе. Поскольку миелин страдает, деятельность нервной системы также ухудшается.
Большинство случаев болезни Александра развивается в младенчестве и включает разрушение миелина. У детей с этой болезнью увеличенный мозг, они страдают от судорог, скованности в руках и ногах и задержки развития. Иногда симптомы появляются позднее, в детстве или даже во взрослом возрасте, и они включают нарушения речи, затруднения при глотании, судороги и плохую координацию. Со временем аномальные белковые отложения, содержащие GFAP, известные как волокна Розенталя, накапливаются в специализированных клетках мозга, называемых астроцитами, которые поддерживают и питают другие клетки головного и спинного мозга.
С 2011 года ученые изучают механизмы накопления GFAP в надежде найти лекарство или соединение, чтобы помочь пациентам. Избыточное накопление GFAP, нарушает структуру и функции астроцитов, из-за чего плохо работают все другие клетки мозга. Проблемы накопления GFAP в астроцитах были также обнаружены при других заболеваниях, таких как гигантская аксонная нейропатия и опухоли астроцитомы.
Исследование осветило ключевые механизмы, участвующие в неправильном складывании GFAP в клетках, и выявила новые признаки тяжести заболевания. Впервые ученые провели четкое молекулярное различие между детьми, которые умирают молодыми, и людьми, которые живут в течение нескольких десятилетий. Эти данные в перспективе помогут выявить новые препараты, которые позволят заблокировать аномальный белок и люди смогут жить обычной жизнью, только принимая препараты.
Читайте также нашу статью про генетические тесты по ссылке.
Найден способ вылечить смертельную врожденную болезнь мозга
Способ вылечить опасное неврологическое расстройство, болезнь Александера, нашли специалисты Висконсинского университета в Мэдисоне. Подробнее об этом они рассказали в статье в журнале Science Translational Medicine.
Болезнь Александера — врожденное неврологическое расстройство, от которого нет лекарства. Из-за нарушений синтеза белка GFAP этот белок накапливается в нейроглии, вспомогательных клетках нейронов, не давая нейронам получать питательные вещества. В самом белке появляются узелковые образования — волокна Розенталя. Они не дают нервным импульсам проходить по миелиновым волокнам.
Ранее исследователи предположили, что улучшить состояние мышей с болезнью Александера можно с помощью антисмысловых олигонуклеотидов, фрагментов генома, способных остановить синтез того или иного белка, взаимодействуя с его матричной РНК. Однако симптомы болезни у мышей оказались в принципе выражены слабо, поэтому оценить степень улучшения поведения и качества жизни было затруднительно.
Теперь авторы работы протестировали терапию антисмысловыми олигонуклеотидами на крысах с болезнью Александера. Как выяснилось, при лечении до развития симптомов крысы в будущем практически уступают по физическому состоянию своим здоровым сородичам. И, даже если симптомы уже проявились, терапия позволяла резко улучшить состояние животных и даже в некоторой степени обратить вспять изменения в белом веществе.
В случае с людьми отличным результатом будет, если удастся хотя бы просто остановить болезнь, отмечают исследователи. Если же удастся избавить мозг от повреждений, это будет просто великолепно. Однако до испытаний на людях метод ждет дальнейшая проверка на других видах млекопитающих, более близких человеку.
Болезнь александера что это такое
Болезнь Александера (AXD) является первым описанным заболеванием, вызванным преимущественно нарушением функции астроцитов (Hanefeld, 2004; Mignot et al., 2004). Болезнь впервые была описана Александером в 1949 г. у 18-месячного мальчика с макроцефалией.
Нейропатологический признак заболевания представлял собой «палочковидные фусцинофильные тельца» в белом веществе головного мозга, которые в дальнейшем были идентифицированы как волокна Розенталя. Глиальный фибриллярный кислый белок (GFAP) присутствует в волокнах Розенталя в высокой концентрации и используется в качестве гистологического маркера.
Мутации гена GFAP (хромосома 17q21) у пациентов с болезнью Александера в дальнейшем были идентифицированы как причина аутосомно-рецессивного заболевания (Brenner et al., 2001).
В соответствии с клинической картиной выделяют три различных типа болезни Александера: младенческий, ювенильный и взрослый.
Наиболее распространенный младенческий тип относится к группе макроцефалических болезней белого вещества. У больных детей отмечается нормальный размер головы при рождении, а затем развивается медленно прогрессирующая мегалэнцефалия в сочетании со спастичностью, раздражимостью и эпилептическими припадками (Pridmore et al, 1993).
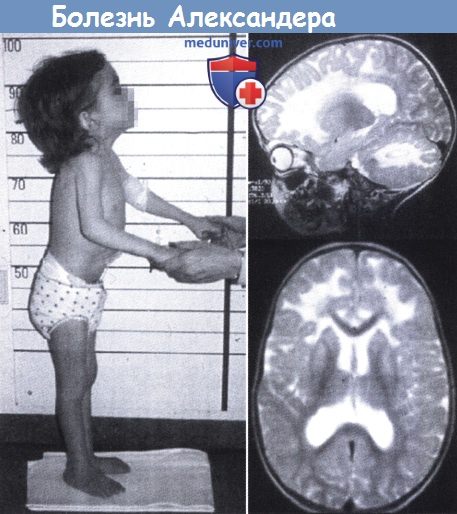 Болезнь Александера. Макроцефалия у четырехлетней девочки (слева).
Болезнь Александера. Макроцефалия у четырехлетней девочки (слева).
На МРТ в сагиттальной проекции в Т2-режиме (справа) определяется повышение сигнала переднего белого вещества; заметно поражение мозжечка.
У пациентов старшего возраста чаще отмечается нормоцефалия. Иногда симптомы ошибочно диагностируются как новообразование ствола мозга. Описаны единичные семьи с несколькими больными, а также монозиготные близнецы (Meins et al., 2002). Заболевание было описано Mignot et al. (2004).
Диагностика классических случаев AXD основана на наличии макроцефалии, регрессии, припадков и бульбарных симптомов у пациентов старшего возраста.
На ЭЭГ можно выявить генерализованную медленную активность и некоторое количество очаговых разрядов. Наиболее ценную информацию можно получить по результатам КТ и МРТ. Описаны нейрорадиологические характеристики, которые практически все гда встречаются при AXD (Van der Knaap et al., 2001). Описанные признаки включают экстенсивное усиление сигнала мозгового белого вещества с преобладанием изменений в лобной области, перивентрикулярный ободок высокого сигнала в Т1-режиме и низкого сигнала в Т2-режиме, аномалии базальных ганглиев, таламуса и ствола мозга, а также усиление контрастирования отдельных структур серого и белого вещества.
При МР-спектроскопии выявляется выраженное накопление миоинозитола и снижение содержания α-нафтилуксусной кислоты, сопровождающееся накоплением лактата в пораженном белом веществе головного мозга и мозжечка. Уровень миоинозитола повышен в белом веществе, а также в сером веществе и базальных ганглиях.
Изменения отражают глиальную (астроцитарную) пролиферацию, а также активную демиелинизацию и нейроаксональную дегенерацию. Совсем недавно описано повышение GFAP в спинномозговой жидкости при всех трех подтипах AXD (Kyllerman et al., 2005). Диагноз подтверждается на основании молекулярно-генетической оценки гена GFAP.
В настоящее время проводится только симптоматическое лечение.
— Вернуться в оглавление раздела «Неврология.»
Редактор: Искандер Милевски. Дата публикации: 16.12.2018
Болезнь Александера

Полезное
Смотреть что такое «Болезнь Александера» в других словарях:
Александера болезнь — 1) (В. Alexander, совр. амер. врач) см. Гипопроконвертинемия; 2) (W. S. Alexander, совр. англ. невропатолог; син. Розенталя лейкодистрофия) наследственная болезнь ц. н. с., характеризующаяся распадом миелина, клинически проявляющаяся судорогами,… … Большой медицинский словарь
Александера болезнь — (Alexander, 1949) передающееся по аутосомно рецессивному типу наследования заболевание преимущественно детей раннего возраста. Симптомокомплекс расстройства образуют следующие основные признаки: 1. высокая температура; 2. судорожные припадки; 3.… … Энциклопедический словарь по психологии и педагогике
Александера болезнь — (Alexander W.S., 1949). Наследственное заболевание, проявляющееся в раннем детском возрасте. Тип передачи – аутосомно рецессивный. Характерны нарушения метаболизма в астроцитах. Патологоанатомически – демиелинизирующая лейкодистрофия. Клинически… … Толковый словарь психиатрических терминов
НЕДОСТАТОЧНОСТЬ ПЛАЗМЕННЫХ ФАКТОРОВ СВЁРТЫВАНИЯ — мед. Плазменные факторы свёртывания крови различные компоненты плазмы, реализующие образование сгустка крови. Недостаточность плазменных факторов свёртывания может быть изолированной или комбинированной. • Изолированная недостаточность • Фактор I … Справочник по болезням
Список наследственных заболеваний — Список генетических заболеваний Основные статьи: наследственные заболевания, Наследственные болезни обмена веществ, Ферментопатия. В большинстве случаев приведен также код, указывающий на тип мутации и связанные с ней хромосомы. См. также система … Википедия
ОЛИГОФРЕНИЯ — – группа различных по этиологии и патогенезу заболеваний, основным проявлением которых служит врожденное или приобретенное в первые 3 года жизни слабоумие и затруднение социальной адаптации. Причиной олигофрении могут быть хромосомные аномалии,… … Энциклопедический словарь по психологии и педагогике
Нарушения кровообращения — (гемодисциркуляторные процессы) типовые патологические процессы, обусловленные изменением объёма крови в сосудистом русле, её реологических свойств или выходом крови за пределы сосудов. Содержание 1 Классификация 2 Гиперемия (полнокровие) … Википедия
Лейкодистрофи́и — (греч. leukos белый + Дистрофия группа наследственных заболеваний, характеризующихся прогрессирующей дистрофией белого вещества головного и спинного мозга. В основе патогенеза Л. лежит генетически детерминированный дефект обмена липидов, миелина … Медицинская энциклопедия
Лейкодистрофия Розенталя — См.: Болезнь Александера … Энциклопедический словарь по психологии и педагогике
ЧИКАГСКАЯ ШКОЛА — Разрабатывает психоаналитический подход к психосоматическим расстройствам, подчеркивая значение эмоциональных факторов в возникновении соматического заболевания и особую роль в создании терапевтической программы. Термин… … Психотерапевтическая энциклопедия