Талассемия
Название «талассемия» происходит от греческого слова «море», «thalassa», и переводится как «морская анемия», так как болезнь впервые была обнаружена у лиц, живущих у берегов Средиземного моря.
Глобин образует гемоглобин. Гемоглобин жизненно важен в организме, так как он входит в состав эритроцитов (красных кровяных клеток), которые переносят кислород от легких ко всем тканям и органам. Талассемия проявляется гемолитической анемией различной степени выраженности. Из-за разрушения неправильного гемоглобина эритроциты быстро разрушаются. Организм пытается скомпенсировать дефицит гемоглобина в организме и усиливает образование красных клеток крови в костном мозге.
Альфа-талассемия – результат недостаточного образования альфа-глобиновых цепей. Бета-талассемия – результат недостаточного синтеза бета-глобиновых цепей.
Клинические проявления талассемии
Легкие, клинически незначимые формы талассемии обычно представлены у гетерозигот, большинство которых являются здоровыми носителями этих заболеваний. В мазках крови этих людей специалисты могут увидеть изменение внешнего вида эритроцитов (красных кровяных клеток) в виде гипохромии (эритроциты более бледные, чем обычно), микроцитоза (уменьшение размера), мишеневид- ности, наличие клеток различной формы (пойкилоцитоз) и размера (анизоцитоз).
Клинически значимые формы талассемии включают следующие состояния:
Большая форма бета-талассемии (анемия Кули), которая проявляется тяжелой анемией (глубоким снижением гемоглобина) уже на первом-втором году жизни и необходимостью проведения переливаний донорских эритроцитов каждые 2-4 недели.
Промежуточная форма бета-талассемии, которая развивается в результате нали- чия генетических повреждений двух бета-глобиновых генов, наличие комбина- ции талассемического повреждения одного бета-глобинового гена с аномальным гемоглобином (например, HbE) или повреждение двух бета-глобиновых генов с нарушением работы альфа-глобиновых генов, или появление аномального HbE. Как правило, пациенты с промежуточной формой бета-талассемии не нуждаются в регулярных (частых) переливаниях донорских эритроцитов.
Гемоглобинопатия Н, которая имеет клинические проявления сходные с промежуточной формой бета-талассемии.
Считается, что промежуточная форма бета-талассемии (а также HbE-бета-талассемия, гемоглобинопатия HbE и гемоглобинопатия Н) в целом более легкая форма талассемии, чем большая форма бета-талассемии, т.к. они редко нуждаются в заместительных трансфузиях (переливаниях) донорских эритроцитов, но в не- которых случаях частота переливаний у них может учащаться: с возрастом, при развитии выраженного увеличения размеров селезенки (спленомегалии), при присоединении инфекций и во время беременности.
Какие проявления талассемии?
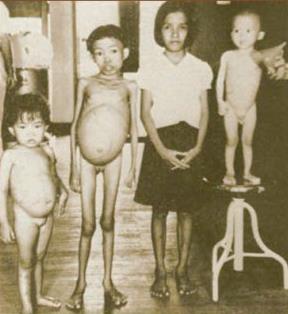
Фото 1. Дети, страдающие средиземноморской анемией тяжелой степени (последующие названия – анемия Кули, большая форма бета-талассемии) из публикации Cooley T.B. & Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans Am Pediatr Soc, 1925; 37: 29-30
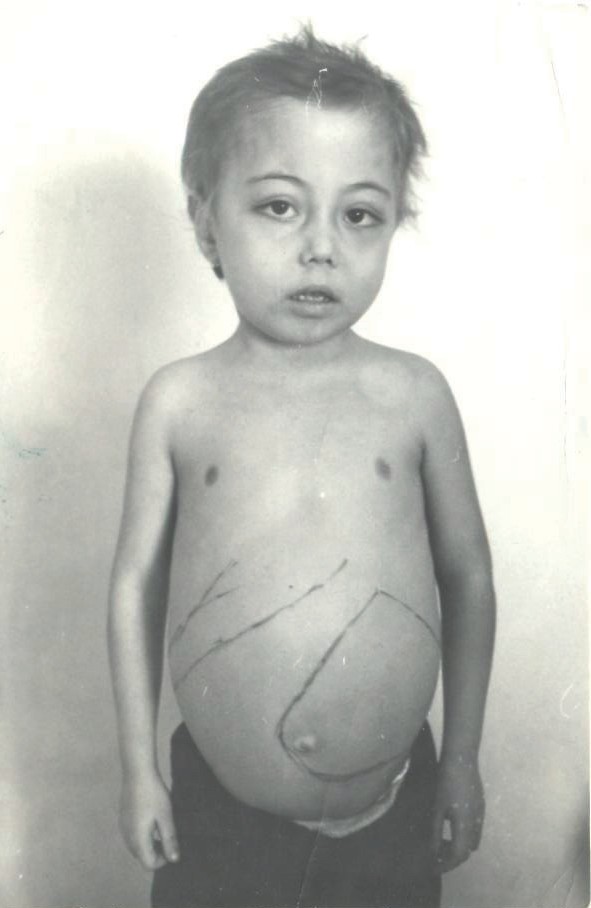
Фото 2. Внешний вид больного большой формой бета-талассемии 12-ти лет, не получав- шего лечение (60-е годы СССР).
Анемия – снижение гемоглобина – результат неэффективного образования эритроцитов в костном мозге и сокращения продолжительности жизни эритро- цитов, в тяжелых случаях анемия настолько выражена, что необходимо посто- янно проводить переливания донорских эритроцитов. Проявляется слабостью, одышкой, желтухой, интоксикацией и повышением температуры тела.
Перегрузка железом патологическое накопление железа в различных органах и тканях, результат переработки донорских эритроцитов организмом пациента в сочетании с резко повышенным усвоением железа из пищевых продуктов; последнее становится основной причиной накопления излишка железа у пациентов с промежуточной формой бета-талассемии. Накопленное железо повреждает печень, сердце и эндокринные железы и они не могут нормально работать.
Увеличение размеров селезенки (спленомегалия) различной выраженности. Увеличение размеров селезенки при сбалансированном ведении больных большой формой талассемии в настоящее время встречается редко и выражено незначительно. При неадекватном лечении селезенка быстро увеличивается в размерах за счет участия в кроветворении (начинает помогать костному мозгу образовывать эритроциты, но общий гемоглобин не повышается). Спленомегалия приводит к снижению эффективности проводимых переливаний донорских эритроцитов, а также к снижению численности других клеток периферической крови – тромбоцитов и лейкоцитов, такое состояние называется гиперспленизмом. При резком выраженном увеличении селезенки появляется высокий риск разрыва селезенки при малейшей травме живота, что представляет угрозу жизни. При промежуточной форме бета-талассемии спленомегалия – частое явление. Очень важно регулярно следить за размерами селезенки, особенно у пациентов с редкими трансфузиями донорских эритроцитов!
Экспансия кроветворения – расширение участков кроветворения в костном мозге. В норме кроветворение располагается в губчатых (плоские кости черепа, ребра, тела позвонков, кости таза) костях и трубчатых костях, с возрастом по мере старения организма – только в плоских костях. При возрастании потребностей организма в клетках крови, как например, при талассемии (промежуточной форме и при неадекватном лечении большой формы) высокая необходимость в эритроцитах, кроветворение занимает все возможные места естественной локализации кроветворения, приводя к утолщению и деформации костей. При экстремальном расширении кроветворения – клетки костного мозга выходят за пределы костей, образовывая опухолевидные образования – псевдоопухоли. За счет костных деформаций и утолщений, а также за счет псевдоопухоли, могут сдавливаться жизненно важные сосудистые и нервные сплетения, что может проявляться различными неврологическими симптомами (потеря чувствительности, чувство онемения, боли, нарушение функции мышц/органов, которые питаются из этого сплетения, и др.), симптомами сдавления (когда псевдоопухоль сильно сдавливает нормальные структуры, мешая их функции), остеопороз, патологические переломы, что также может сопровождаться болью и нарушением функции организма.
При проведении недостаточной трансфузионной терапии развиваются типичные талассемические деформации костей скелета увеличение размеров живота за счет спленомегалии, обусловленные расширением кроветворения, замедляются темпы физического роста и развития за счет глубокой анемии. Внешний вид боль- ных талассемией, не получающих лечение, представлен на фото 1 и 2.
Исследования для подтверждения талассемии
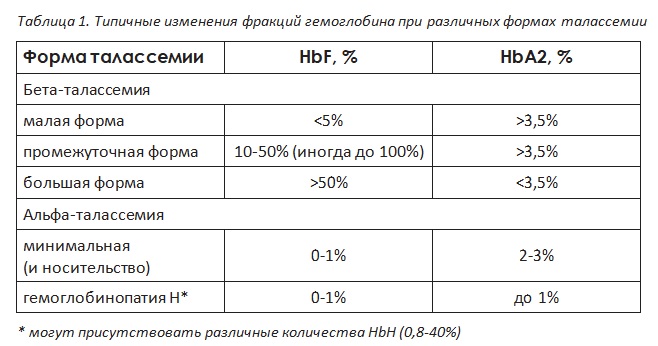
Исследование фракций гемоглобина методом капиллярного электрофореза или высокоэффективной жидкостной хроматографии. В случае талассемии будут изменены пропорции нормальных фракций гемоглобина или выявлен аномальный гемоглобин (см. таблицу 1).
ДНК-исследование глобиновых генов генов позволяет выявить как точковые мутации, так и делеции (потери фрагментов гена) различной протяженности. Знание характера мутации позволяет предположить возможную тяжесть заболевания. Часть поломок бета-глобинового гена позволяет сохраниться некоторому остаточному синтезу бета-глобиновых цепей, другая часть – делает синтез этих цепей полностью невозможным. Необходимо исследовать как бета-глобиновый ген, так и альфа-глобиновые гены в связи с крае высокой частотой встречаемости сочетанных поражений. Сочетание альфа- и бета-талассемии приводит к некоторому выравниванию баланса глобиновых цепей и смягчению клинических проявлений заболевания.
Ультразвуковое исследование (УЗИ) органов брюшной полости необходимо для оценки размеров печени и селезенки, состоятельности желчевыводящих путей.
Стандартная терапия талассемии
Больные малой формой бета-талассемии и здоровые носители талассемии не требуют лечения. Для них очень важно соблюдение здорового образа жизни : сбалансированное питание, двигательная активность, отсутствие вредных привычек (курение, алкоголь, употребление токсичных веществ), что позволит организму самостоятельно компенсировать генетический дефект.
Больные тяжелыми клиническими формами талассемии нуждаются не только в здоровом образе жизни, но и в специальном лечении.
Для всех пациентов с любой формой талассемии в любом возрасте принципиально важна двигательная активность – занятия физкультурой и спортом (без участия в соревнованиях), что позволит максимально поддержать минерализацию костей скелета, особенно при средне тяжелых и тяжелых клинических формах талассемии.
Трансфузионная терапия (переливания эритроцитной массы)
Пациенты с промежуточной формой бета-талассемии (и другими клинически схожими формами талассемии) в трансфузиях донорских эритроцитов нуждают- ся редко (например, при снижении гемоглобина менее 50 г/л, при сопутствующих инфекциях, при проведении хирургических операций, на время беременности, при выраженной задержке роста, легочной гипертензии, сердечной недостаточности, склонности к тромбозам, тяжелые костные деформации).
Пациенты с большой формой бета-талассемии для сохранения нормальных темпов роста и развития в детском возрасте, для сохранения качества и продолжительности жизни не отличающегося от здоровых людей – во взрослом возрасте, вынуждены регулярно каждые 3-4 недели в течение всей жизни получать трансфузии (переливания) донорских эритроцитов.
При назначении переливаний донорских эритроцитов врач подробно объяснит как будет проходить трансфузия, почему ее нужно проводить, проведет пробу на совместимость донорских эритроцитов с вашей кровью, объяснит какие могут быть реакции на переливание и что нужно вам делать в этих случаях. Необходимо внимательно выслушать доктора, если у вас возникнут какие-либо вопросы, не стесняйтесь, задайте их до начала трансфузии, и далее тщательно следуйте советам врача.
Это лечение, направленное на удаление избытка железа из организма пациента. Используются лекарственные препараты, разрешенные к медицинскому применению на территории Российской Федерации (деферазирокс); в случае непереносимости пациентом этих препаратов, решением Врачебной комиссии с соблюдением всех регламентируемых Российским законодательством норм может быть назначен препарат, незарегистрированный в России, но разрешенный в других странах мира для лечения перегрузки железом у больных талассемией (дефероксамин).
Исследования, необходимые для контроля лечения
Общий анализ крови с подсчетом числа тромбоцитов, лейкоцитарной формулы, ретикулоцитов и нормобластов (молодые ядросодержащие клетки красной крови, в норме отсутствуют в периферической крови), который позволит врачу оценить насколько адекватно организовано лечение клинически значимой формы талассемии.
Биохимический анализ крови с оценкой функции печени и почек, запасов железа в организме (ферритин сыворотки) позволяет оценить безопасность проводимой вам (вашему ребенку) терапии.
УЗИ органов брюшной полости и забрюшинного пространства – необходимо для оценки размеров печени, селезенки и почек, ранее выявление камней в желчном пузыре и псевдоопухолевых масс внекостномозгового кроветворения
Исследование функции сердца (ЭХО-КГ и ЭКГ, при необходимости суточное мониторирование ЭКГ) для всех пациентов с клинически значимыми формами талассемии с момента постановки диагноза и далее ежегодно.
Исследование гормонального профиля для всех трансфузионно зависимых пациентов старше 8 лет.
Исследование содержание железа в печени, миокарде и гипофизе до начала хелаторной терапии и для контроля освобождения органов от избытка железа.
Скрининг на инфекции, передающиеся с компонентами донорской крови (ВИЧ, гепатиты В и С) случае регулярных трансфузий – ежемесячно, при редких трансфузиях – ежегодно.
Оценка остроты зрения и слуха до начала хелаторной терапии и далее ежегодно.
Оценка минеральной плотности костей скелета ежегодно начиная с подросткового возраста.
Радикальная терапия талассемии
В настоящее время радикальной терапией клинически значимых форм талассе- мии считается трансплантация гемопоэтических стволовых клеток (ТГСК), источником которых может быть костный мозг, периферическая кровь и пуповинная кровь. Общая бессобытийная выживаемость больных талассемией после ТГСК достигает 98% при условии, что до этого больной получал адекватную терапию (трансфузии донорских эритроцитов при гемоглобине 95-100 г/л до Hb не менее 125 г/л в сочетании с хелаторной терапией (на фоне проводимой хелации ферритин сыворотки менее 1500 мкг/л), незначительное увеличение селезенки (не выступает ниже края реберной дуги), отсутствие признаков фиброза в печени.
В противном случае общая бессобытийная выживаемость – существенно ниже, а при тяжелой перегрузке железом печени и миокарда, наличии анемической кардиомиопатии, выраженной спленомегалии – не целесообразна в связи с крайне высоким риском гибели пациента.
В последние годы появилась информация по генной терапии, которая скоро сможет стать еще одной радикальной опцией лечения.
Что делать, если установлен диагноз «талассемия»?
Ваш лечащий врач должен отправить вас на консультацию к местному гематологу, который затем, должен дать направление в один из Федеральных центров, указанных ниже, для проведения диагностики (подтверждения талассемии) и назначения правильного лечения.
Для получения необходимого лечения также важно оформить инвалидность, поэтому в заключении лечебного учреждения, кроме информации о терапии, должны быть рекомендации о прохождении МСЭ по месту жительства.
В соответствии с Перечнем групп населения и категорий заболеваний, при амбулаторном лечении которых, лекарственные средства и изделия медицинского назначения отпускаются по рецептам врачей бесплатно, утвержденном постановлением Правительства РФ № 890 от 30.07.1994 «О государственной поддержке развития медицинской промышленности и улучшении обеспечения населения и учреждений здравоохранения лекарственными средствами и изделиями медицинского назначения», дети-инвалиды в возрасте до 18 лет обеспечиваются всеми лекарственными средствами по рецептам врачей бесплатно.
Кроме того, талассемия относится к гематологическим заболеваниям (наследственным гемопатиям), и препараты для лечения данных заболеваний и коррекции осложнений их лечения также должны предоставляться пациентам по рецептам врачей бесплатно.
ФЕДЕРАЛЬНЫЕ ЦЕНТРЫ, ОКАЗЫВАЮЩИЕ СПЕЦИАЛИЗИРОВАННУЮ ПОМОЩЬ ПАЦИЕНТАМ С ТАЛАССЕМИЕЙ
1. ФГБУ «Национальный медицинский исследовательский центр детской гема- тологии, онкологии и иммунологии имени Дмитрия Рогачева» Минздрава России (Москва)
Адрес: г. Москва, ул. Саморы Машела, д. 1
2. ФГБУ Национальный медицинский центр гематологиии МЗ РФ
Адрес: г. Москва, Новый Зыковский проезд, д. 4а
ФГБОУ ВО «Первый Санкт-Петербургский государственный медицинский университет имени академика И.П. Павлова министерства здравоохранения Российской Федерации», клиника НИИДОГиТ им. Р.М. Горбачевой.
Адрес: г. Санкт-Петербург, ул. Рентгена, д.12
Федеральное государственное бюджетное научное учреждение «Медико-генетический научный центр»
Адрес: г. Москва, ул. Москворечье, д. 1
Часто задаваемые вопросы
Наиболее вероятно у Вашего ребенка – талассемия, поэтому надо провести специальное обследование у гематолога (исследование фракций гемоглобина, ДНК-исследование глобиновых генов). После уточнения формы талассемии можно будет сказать как часто в дальнейшем надо переливать «кровь». Переливания необходимы для нормального роста и развития ребенка.
Задержка роста у ребенка с бета-талассемией, получающего переливания, может быть связана с двумя причинами:
— в результате переливаний донорских эритроцитов и/или длительно существующей глубокой анемии (Hb менее 90 г/л) происходит накопление излишка железа, которое нарушает рост костей; для устранения этой проблемы необходимо исследовать обмен железа и начать хелаторную терапию (выведение избытка железа из организма) или откоррегировать дозу препарата – хелатора железа.
При тяжелых формах бета-талассемии (промежуточная и большая формы) у больных нарушается обмен кальция, развивается остеопения и даже остеопороз, что сопровождается в том числе нарушением минерализации зубов. Необходимо провести исследования крови на содержание витамина Д и кальция в крови, после чего врач назначит необходимое лечение.
Вероятность рождения в семье второго больного ребенка составляет 25%, т.к. оба родителя являются носителями заболевания. В настоящее время в России можно провести пренатальную диагностику бета-талассемии и предотвратить рождение больных детей в семье. Для получения более подробной информации по пренатальной диагностике необходимо обратиться к генетику.
Пациентам с малой формой бета-талассемии при условии ведения здорового образа жизни ничего не угрожает, дети – развиваются как все здоровые дети, а взрослые имеют продолжительность и качество жизни как все остальные люди. При этом в общем анализе крови будут сохраняться типичные для заболевания изменения. Если нарушается питание (в рационе недостаточно свежих овощей и фруктов и/или мяса (рыбы)), то гемоглобин в крови будет ниже обычного; при малоподвижном образе жизни – нарушится формирование костей; при употреблении алкоголя (тоников) может развиться токсический гепатит в более раннем возрасте нежели у людей без бета-талассемии.
Больные большой формой бета-талассемии, получающие полноценное (адекватное) лечение – гемоглобин сохраняется в диапазоне 100-125 г/л, ферритин сыворотки всегда менее 1000 мкг/л, растут и развиваются как все здоровые дети и подростки, т.е. пубертат (половое развитие) наступит во время и репродуктивная функция страдать не будет. У таких пациенток может наступить беременность, которая благополучно завершиться
В случае не адекватного лечения половое развитие будет существенно отставать, а в результате избытка железа произойдет необратимое нарушение функции гипофиза, что сопровождается неизлечимым нарушением репродукции. Это значит, что самостоятельно беременность не наступит.
Девушки с большой формой бета-талассемии могут иметь клинически здоровых детей, т.к. они будут носителями бета-талассемии. Необходимо обследовать жениха на предмет носительства талассемии. Если он – носитель талассемии, то обязательное проведение пренатальной диагностики. За более подробной информацией вам нужно обратиться к генетику.
Трансплантация гемопоэтических стволовых клеток может быть проведена только при соблюдении следующих условий:
— нет увеличения печени и селезенки;
— нормально функционирует сердце;
— нет выраженной перегрузки организма железом;
— нет фиброза печени;
— возраст младше 14 лет;
— найден совместимый донор.
Если все условия соблюдены, то вероятность успешного завершения составляет более 95%.
Как правило, к 2-м годам больные с большой формой бета-талассемии и к примерно 7 годам с промежуточной формой бета-талассемии накапливают существенный избыток железа. Ферритин сыворотки при этом будет более 1000 мкг/л при большой форме и более 600 мкг/л при промежуточной форме. Желательно проводить дополнительное обследование – МРТ в режиме Т2* с расчетом накопления железа как в печени, так ив сердце, что позволит более точно подобрать правильную дозу препарата.
В настоящее время для выведения избытка железа в России есть два препарата – Эксиджад и Джадену. Оба препарата имеют одинаковую эффективность, отличаются лекарственной формой: Эксиджад – таблетки диспергируемые (т.е. перед употреблением их необходимо растворить как написано в инструкции), а Джадену – таблетки, покрытые кишечно-растворимой оболочкой (т.е. таблетку можно проглотить целиком или смешать с нежирной пищей после измельчения).
Не реже одного раза в год, частоту и объем обследования назначает ваш гематолог.
Врачи на основании клинических симптомов могут заподозрить болезнь. Затем проводятся лабораторные тесты и инструментальное исследование. СГХС обычно первоначально распознается по аномально высоким уровням ХС ЛПНП, определяемым по данным теста на холестерин. Кроме того, для подтверждения диагноза может быть выполнено генетическое тестирование. В анализе крови (развернутая липидограма), как правило, выявляют: повышение уровня общего холестерина, липопротеидов низкой плотности (ЛПНП), нормальный уровень липопротеидов высокой плотности (ЛПВП) и триглицеридов.

Семьям очень важно посетить врача генетика. Лечащие врачи не всегда имеют возможность поговорить с семьей и рассказать о всех рисках и возможностях, которые может предоставить современная генетика. У врача генетика можно узнать риск рождения больного ребенка в данной семье, пройти обследование родственникам, если это необходимо.
Где в России занимаются диагностикой и лечением можно прочитать в разделе КОНТАКТЫ
Талассемия
Талассемия – это генетическая болезнь крови, при которой, из-за мутации генов, образовывается недостаточное количество гемоглобина в организме и происходит деформация эритроцитов.

Гемоглобин – это специфический белок, который содержится в эритроцитах и отвечает за перенос железа и кислорода ко всем органам и тканям. Основная функция гемоглобина – дыхательная и его недостаток приводит к кислородному голоданию. В результате чего органы не могут полноценно выполнять свои физиологические функции.
Различают «взрослый» гемоглобин (HbA) и плодный тип гемоглобина (HbF).
Фетальный (плодный) гемоглобин синтезируется у плода спустя 2 недели после формирования внутренних органов (приблизительно с 12-й недели беременности). После рождения ребенка, HbF составляет до 80% и постепенно замещается «взрослым» HbA.
В норме у человека, плодного типа гемоглобина должно остаться не более 1,5%. Если случился сдвиг цифр влево или вправо – это всегда говорит о патологии (может быть нарушена работа сердечно-сосудистой системы, щитовидной железы, гипофиза, наличие гематологических отклонений, лейкоза, лимфогранулематоза, интоксикации).
Здоровая зрелая молекула гемоглобина (HbA) состоит из двух пар цепей – альфы и беты. Фетальный гемоглобин (HbF) состоит из цепей – гаммы и дельты.
А талассемия – это результат нарушенного синтеза одной или нескольких цепей гемоглобина. На основе чего различают альфа-талассемию, бета-талассемию, бета-дельту талассемию и т.д.
Тяжесть протекания того или другого вида заболевания зависит от количества поврежденных участков:
Большая альфа-талассемия – самое серьезное отклонение, которое возникает еще в период внутриутробного развития плода. Грозит осложнениями, как для ребенка, так и для женщины.
Сегодня при своевременной внутриутробной трансфузии эритроцитарной массы удается сохранить жизнь плода.
Большая бета-талассемия (анемия Кули) – это также опасное нарушение, которое проявляется в первые два года жизни ребенка.
Малая альфа и бета-талассемия нарушает процесс эритропоэза (развитие новых эритроцитов), что приводит к хронической анемии и нарушению гемолиза.
Вне зависимости от вида, клиническое протекание талассемий схожи, различия зависят от степени тяжести патологического процесса.
Причины
Главная причина талассемии – наследственный фактор. При котором у одного пациента функционирует патологическая РНК (участвует в кодировании, чтении и регуляции генов), а у другого – ДНК (появляются изменения в хромосоме). На фоне этих изменений уменьшается или полностью прекращается синтез одной из цепочек Hb.
В норме количество альфа, бета-цепей – одинаково, поэтому изменение синтеза хотя бы одной из них приводит к дисбалансу и патологии.
Болезнь встречается как у девочек, так и у мальчиков. Частота появления большой талассемии 1:100 000, если оба родители носители мутационного гена.

Также на патогенез талассемии влияет и этническая принадлежность, так как чаще всего данная генетическая патология встречается у жителей Африки, Восточного Средиземноморья и Юго-Восточной, Центральной Азии. Определено это тем, что мутационный ген имеет большую сопротивляемость к малярии (обычно в африканских регионах) и большим количеством родственных браков, что приводит к разным генетическим нарушениям, включая талассемию.
Всемирная организация здравоохранения приняла международную классификацию заболеваний 10-го пересмотра ( мкб-10 ), как единый нормативный документ для ведения учета болезней, причин обращений пациентов в медицинские учреждения и причин смертности.
Согласно мкб талассемии (D56) имеет следующую классификацию:
Обновление кодов (мкб-11) будет вводиться в действие с 2022 года.
Симптомы
Талассемия определяется до рождения ребенка при помощи пренатального скрининга.
Если на данном этапе болезнь не была выявлена, следует ожидать такие симптомы:
По результатам лабораторных анализов:
По результатам УЗИ диагностики:
Проявление первых симптомов талассемии зависит от вида и количества «деформированных» генов.
Симптомы большой альфа-талассемии проявляются сразу после рождения, большой бета-талассемии – в первые два года жизни малыша.
Поэтому, бета-талассемия может быть дополнена следующими признаками:
Включая симптомы сердечно-сосудистых, эндокринных патологий и заболеваний печени.
При талассемии резко увеличивается усвоение железа из продуктов питания – это приводит к дисбалансу работы внутренних органов и систем. При отсутствии корректной диагностики и лечения талассемии – остается высокий риск летального исхода в первый год жизни.
Основные нарушения в организме, к которым приводит талассемия:
Если у пациента поражен только один ген – талассемия не имеет симптоматических проявлений. Однако следить за уровнем гемоглобина и размером селезенки – необходимо.
Диагностика
Так как симптоматика талассемии не всегда бывает специфической — врач для постановки окончательного диагноза, в большей мере опирается на результаты лабораторных исследований крови и спинномозговой жидкости.
Поэтому, при подозрении на талассемию необходимо:
Во время исследования крови обращают внимание:
При исследовании костного мозга:
УЗИ и рентгенография нацелены:
Для того чтобы предотвратить негативные последствия болезни, родителям рекомендуется в профилактических целях в период беременности пройти внутриутробный скрининг плода на определение разных генетических патологий, включая талассемию.
Существуют два способа пренатальной диагностики:
Что позволит своевременно определить заболевание и предпринять все необходимые меры.
Лечени
Малая талассемия (нарушение одного гена) – не требует лечения. Достаточно здорового образа жизни, сбалансированного питания, физической активности и отсутствия пагубных привычек. Все это «поможет» организму самостоятельно компенсировать дефект гена.

Большая талассемия и средней степени тяжести нуждается в специальном лечении, которое в «Клинике Спиженко» включает:
Лекарственные препараты при талассемии назначаются только для коррекции симптомов и предупреждения осложнений. Медикаментозного лечения талассемии – нет.
Пациенты с талассемией средней тяжести в донорских трансфузиях нуждаются не часто, только в критических ситуациях для организма:
Большая талассемия требует более серьезного лечения:
Удаление селезенки уменьшает потребность в гемотрансфузиях. Однако отсутствие природного «фильтра» увеличивает риск возникновения разнообразных инфекций, что может негативно отразиться на самочувствии пациента.
Единственным, но радикальным вариантом лечения тяжелых пациентов с талассемией, остается пересадка костного мозга.
Данная терапия позволит и детям и взрослым с талассемией вести нормальный образ жизни с максимальным сохранением ее качества.
Отсутствие должного лечения большой бета-талассемии приводит к ранней смертности пациентов
Для контроля результатов лечения талассемии назначается:
Включая изучение гормонального фона (для пациентов с 8 лет), минеральной плотности костей, остроты зрения и слуха.
Правильный образ жизни и серьезный подход к лечению при талассемии средней тяжести не представляет собой угрозы для нормальной жизнедеятельности пациента. Поэтому прогноз протекания болезни остается достаточно оптимистичным.
Чего нельзя сказать о тяжелой талассемии. Без должного комплексного лечения уменьшается не только продолжительность жизни, но и ее качество.
Поэтому, тем людям, у которых в семье есть родственники с данным наследственным отклонением, нужно пройти комплексное генетическое исследование в обязательном порядке. Особенно в том случае, если вы планируете рождение ребенка.
Заполните всего три поля
и наш специалист Вам перезвонит!
Ваши данные успешно отправлены! Ожидайте нашего звонка.