Клинические рекомендации по диагностике и лечению системного амилоидоза
В клинических рекомендациях, подготовленных специалистами различного профиля, рассматриваются методы диагностики и лечениясистемного амилоидоза, в том числе АА (вто-ричный амилоидоз при хронических воспалительных заболеваниях, включая ревматоидныйартрит, анкилозирующий спондилит, аутовоспалительные заболевания, хроническиенагноения, злокачественные опухоли и др.), AL (амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема) иATTR (транстиретиновый; семейные формыполиневропатического, кардиопатического идругого амилоидоза, системный старческийамилоидоз). Диагноз амилоидоза, которыйможно заподозрить на основании клиническихданных, необходимо подтвердить при гистологическом исследовании (окрашивание препаратов ткани конго-красным с последующей микроскопией в поляризованном свете). Чтобы замедлить или приостановить прогрессирование амилоидоза любого типа, необходимо добиться уменьшения количества (или, если возможно, удаления) белков-предшественников путем лечения хронического воспаленияпри АА-амилоидозе или подавления пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуно-глобулинов при AL-амилоидозе. Для замедления прогресирования ATTR-амилоидоза упациентов с полиневропатией применяют тафамидис, который ингибирует диссоциацию мутантного транстиретина и снижает его амилоидогенность.
Определение, классификация, группы риска и принципы диагностики
Амилоидоз – группа заболеваний, отличительным признаком которых является отложение в тканях и органах фибриллярного гликопротеида амилоида. Специфическое свойство амилоида, отличающее его от других фибриллярных белков стромы, – способность к двойному лучепреломлению, что проявляется свечением в поляризованном свете предварительно окрашенных конгокрасным препаратов амилоида с изменением красного цвета конгофильных амилоидных отложений на яблочно-зеленый (дихроизм).
В основе амилоидогенеза лежит синтез большого количества нестабильных белковпредшественников, которые агрегируются с образованием амилоидной фибриллы. Клю чевое значение имеет амилоидогенность основного белка-предшественника амилоида, специфичного для каждой формы амилоидоза (в настоящее время известно более 30 таких белков), обозначение которого положено в основу современной классификации заболевания (ВОЗ, 2016 г.). Названия типов амилоида включают в себя букву А, означающую “амилоид», и обозначение конкретного фибриллярного белка амилоида – А (амилоидный А-протеин), L (легкие цепи иммуноглобулинов), TTR (транстиретин), β2М (β2-микроглобулин), В (В-протеин), IAPP (островковый амилоидный полипептид). Используют также производные наименования – иммуноглобулиновый амилоидоз (AL), транстиретиновый (ATTR) и др. (табл. 1) 3. Следует отметить, что Международная классификация болезней (МКБ) 10-го пересмотра базируется на клиническом принципе, не учитывает особенности патогенеза различных форм амилоидоза и не позволяет обосновать адекватное лечение.
| Белок амилоида | Белок-Белок-предшественник | Клиническая форма амилоидоза |
|---|---|---|
| АА | SSA-белок | Вторичный амилоидоз при хронических воспалительных заболеваниях, в том числе периодической болезни и синдроме Макла-Уэллса |
| AL | λ, κ-легкие цепи иммуноглобулинов | Амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема |
| ATTR | Транстиретин | Семейные формы полиневропатического, кардиопатического и др. амилоидоза, системный старческий амилоидоз |
| Аβ2М | β2-микроглобулин | Диализный амилоидоз |
| AGel | Гелсолин | Финская семейная амилоидная полиневропатия |
| AApoAI | Аполипопротеин А-I | Амилоидная полиневропатия (III тип по van Allen, 1956 г.) |
| AFib | Фибриноген | Амилоидная нефропатия |
| Aβ2 | β-белок | Болезнь Альцгеймера, синдром Дауна, наследственные кровоизлияния в мозг с амилоидозом |
| APrPScr | Прионовый белок | Болезнь Крейтцфельда-Якоба, болезнь Герстманна-Штраусслера-Шейнкера |
| AANF | Предсердный натрийуретический фактор | Изолированный амилоидоз предсердий |
| AIAPP | Амилин | Изолированный амилоидоз в островках Лангерганса при сахарном диабете 2 типа, инсулиноме |
| ACal | Прокальцитонин | При медуллярном раке щитовидной железы |
| ACys | Цистатин С | Наследственные кровоизлияния в мозг с амилоидозом (Исландия) |
АА-амилоидоз чаще всего развивается при ревматоидном артрите, серонегативных спондилоартропатиях, аутовоспалительных наследственных периодических лихорадках, в том числе периодической болезни (семейной средиземноморской лихорадке), а также при хронических нагноениях, туберкулезе. АА-амилоид образуется из сывороточного предшественника SAA (serum amyloid A) – острофазового белка, продуцируемого в значительных количествах в ответ на воспаление. По этой причине АА-амилоидоз называют также реактивным или вторичным.
Клинические формы AL-амилоидоза обусловлены единым этиологическим фактором – В-лимфоцитарной дискразией, характеризующейся формированием аномального клона плазматических или В-клеток в костном мозге, которые продуцируют аномальные иммуноглобулины, обладающие амилоидогенностью (легкие цепи моноклонального иммуноглобулина, чаще λ, реже κ-типа). При первичном AL-амилоидозе плазмоклеточная дискразия относительно более доброкачественная, в то время как при В-гемобластозах (множественной миеломе, болезни Вальденстрема и др.) она обладает признаками злокачественной опухоли. Аномальный амилоидогенный клон плазматических клеток может формироваться также из плазмоцитов, локализующихся вне костного мозга, что может привести к развитию локального амилоидоза. Наиболее распространенные локальные формы AL-амилоидоза – амилоидоз трахеи, бронхов и гортани, мочевого пузыря. Выявление плазмоклеточной дискразии необходимо для диагностики AL-амилоидоза, а также для оценки его риска и дифференциального диагноза.
ATTR-амилоидоз является необратимо прогрессирующим заболеванием с высокой степенью инвалидизации вследствие тяжелого поражения сердца, периферической и/или автономной полиневропатии. Пациенты обычно умирают в течение 10-12 лет от первых проявлений. Развитие ATTR-амилоидоза обусловлено мутациями в молекуле транстиретина или возрастным нарушением секреции тетрамеров транстиретина печенью. В обоих случаях происходит распад тетрамеров транстиретина до мономеров, обладающих выраженной конформационной нестабильностью.
Рекомендации:
Клинические проявления
Для вторичного АА-амилоидоза характерно более раннее начало, чем для AL-амилоидоза (средний возраст больных составляет около 40 и 65 лет, соответственно). ATTR-амилоидоз, несмотря на наследственную природу, характеризуется низкой пенетрантностью и также проявляется обычно после 35 лет.
Поражение почек – ведущий клинический признак АА- и AL-амилоидоза, наблюдающийся практически у всех больных. Поражение почек встречается и у больных с многими формами семейного амилоидоза (AFib, ALys, AGel и др.). При ATTR-амилоидозе нефропатия отмечается лишь у 20-23% больных. Клинически амилоидная нефропатия характеризуется неуклонно прогрессирующим течением с последовательной сменой стадий: протеинурия, нефротический синдром, хроническая почечная недостаточность (ХПН). Иногда возможно развитие ХПН без предшествующего нефротического синдрома.
Поражение сердца развивается у подавляющего большинства больных AL-амилоидозом и у 50-60% пациентов с АTTR-амилоидозом, но не характерно для АА-амилоидоза (рис. 1). При эхокардиографии у больных амилоидозом сердца наблюдается утолщение межжелудочковой перегородки и стенки левого желудочка (чаще симметричное), которое не сопровождается электрокардиографическими признаками гипертрофии миокарда. У части больных отмечается снижение вольтажа зубцов на ЭКГ, хотя отсутствие этого признака не исключает диагноз амилоидоза сердца. Нарушение диастолической функции левого желудочка (рестриктивный тип) приводит к развитию сердечной недостаточности, которая быстро прогрессирует, плохо поддается лечению и почти у 50% пациентов оказывается причиной смерти. Кроме того, у больных амилоидозом сердца часто наблюдаются различные аритмии и нарушения проводимости.
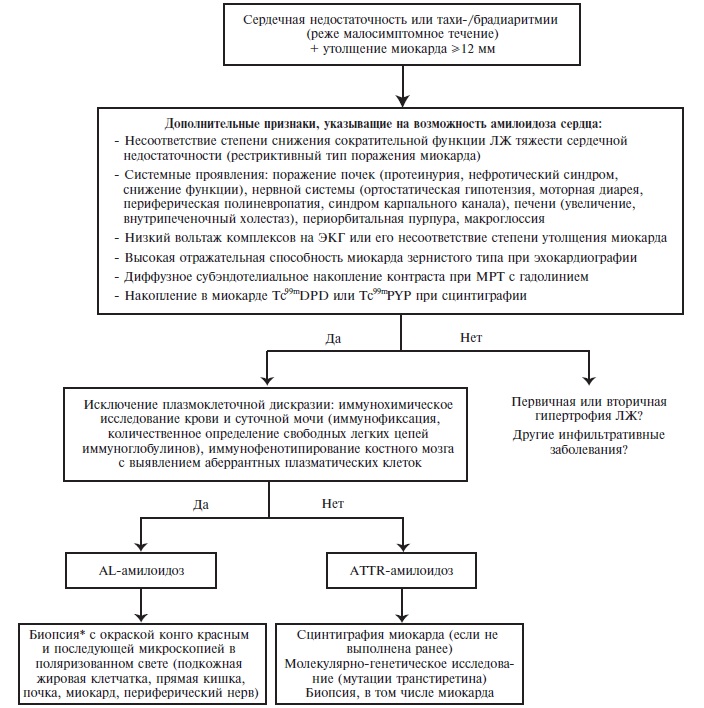 Рис. 1. Алгоритм дифференциальной диагностики амилоидоза сердца. *Если предполагается амилоидоз сердца, то скрининговую биопсию (подкожной жировой клетчатки, слизистой оболочки прямой кишки) целесообразно выполнить параллельно с другими исследованиями. При отрицательном результате показана биопсия пораженного органа, включая сердце.
Рис. 1. Алгоритм дифференциальной диагностики амилоидоза сердца. *Если предполагается амилоидоз сердца, то скрининговую биопсию (подкожной жировой клетчатки, слизистой оболочки прямой кишки) целесообразно выполнить параллельно с другими исследованиями. При отрицательном результате показана биопсия пораженного органа, включая сердце.
При AL-амилоидозе и особенно ATTR-амилоидозе часто встречается ортостатическая артериальная гипотензия – вариант сосудистой недостаточности, при которой сосуды теряют способность поддерживать нормальное артериальное давление в условиях ортостатических нагрузок. Она проявляется ощущением дурноты и потемнением в глазах в ортостазе в сочетании с резким снижением АД. Обычно этот симптом связан с дисфункцией автономной нервной системы (амилоидоз нервных сплетений сосудов). Тяжелая ортостатическая гипотензия сопровождается обмороками, а иногда приводит к развитию острого нарушения мозгового кровообращения.
Поражение желудочно-кишечного тракта может проявляться, особенно при AL-амилоидозе, тяжелой диареей или динамической непроходимостью, которые чаще связаны с нарушениями моторики кишечника вследствие дисфункции автономных нервных сплетений. Иногда выявляют изъязвления или перфорацию стенок с возможным кровотечением. При поражении пищевода возможна дисфагия.
Поражение печени при АА- и AL-типах амилоидоза наблюдают практически в 100% случаев. Функция печени чаще остается сохранной, редким признаком амилоидоза печени является внутрипеченочная портальная гипертензия. При некоторых вариантах семейного ALys-амилоидоза описаны тяжелые спонтанные внутрипеченочные кровотечения.
Увеличение селезенки, обусловленное амилоидным поражением, отмечается у большинства больных и обычно сопутствует увеличению печени.
Поражение нервной системы, представленное симптомами периферической соматической и автономной невропатии, отмечают у 17-35% больных AL-амилоидозом и практически у всех пациентов с наследственной амилоидной полиневропатией разных типов (ATTR, AApoA1 и др.). В большинстве случаев развивается дистальная симметричная полиневропатия с неуклонно прогрессирующим течением, различные дисфункции автономной нервной системы. Реже выявляют двусторонний синдром запястного канала, обусловленный сдавлением срединного нерва депозитами амилоида.
Поражение кожи наблюдают почти у 40% больных AL-амилоидозом. Помимо параорбитальных геморрагий описаны также папулы, бляшки, узелки, пузырьковые высыпания, склеродермоподобная индурация кожи.
Амилоидные отложения в мышцах чаще встречаются при AL-амилоидозе. Макроглоссия – патогномоничный симптом AL-амилоидоза, развивающийся примерно у 20% пациентов.
Редким проявлением амилоидоза, описанным при AL- и, в особенности, АTTR-типах, бывает поражение глаз (сухой кератоконъюнктивит, вторичная глаукома, помутнение стекловидного тела, дисфункции зрачка).
Клиническая картина других типов амилоидоза варьируется в зависимости от основной локализации и распространенности амилоидных депозитов, которые иногда могут быть значительными и напоминать проявления AL-амилоидоза.
Рекомендации:
Лечение системного амилоидоза
Целью терапии любого типа амилоидоза служит уменьшение количества (или, если возможно, удаление) белков-предшественников для того, чтобы замедлить или приостановить прогрессирование болезни. Неблаго приятный прогноз при естественном течении амилоидоза оправдывает применение агрессивных методов лечения. Клиническое улучшение, достигаемое с помощью лечения, включает стабилизацию или восстановление функции жизненно важных органов, а также предотвращение функциональных нарушений с увеличением продолжительности жизни больных. Лечение амилоидоза должно включать симптоматические методы, направленные на уменьшение выраженности сердечной недостаточности, аритмии, отечного синдрома, коррекцию артериальной гипотензии и др.
Лечение АА-амилоидоза
Цель терапии АА-амилоидоза – подавление продукции белка-предшественника SAA (вплоть до устойчивой нормализации), что достигается активным лечением хронического воспаления (в том числе субклинического). Это позволяет уменьшить клинические проявления и предотвратить прогрессирование амилоидной нефропатии и существенно улучшить прогноз.
Рекомендации:
Лечение АL-амилоидоза
При AL-амилоидозе, как и при множественной миеломе, целью лечения служит подавление пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуноглобулинов. В отличие от множественной миеломы, принципиальной задачей лечения AL-амилоидоза является по возможности полная элиминация патологического клона. В связи с быстрым прогрессированием заболевания важное значение имеет применение быстродействующих схем лечения на основе бортезомиба. По мере достижения ремиссии у некоторых больных применяют высокодозную химиотерапию с поддержкой аутологичными стволовыми клетками. При строгом подборе больных с исключением противопоказаний к этой терапии эффект достигают у 60% больных. У больных с клиническими симптомами амилоидоза сердца, ортостатической гипотензией, диареей, желудочно-кишечными кровотечениями в анамнезе, а также у лиц старше 70 лет с амилоидным поражением двух и более систем организма проведение высокодозной химиотерапии не рекомендуется. Тяжелый агранулоцитоз и другие осложнения существенно ограничивают ее применение. Проводят также лечение талидомидом или леналидомидом. Колхицин при AL-амилоидозе не эффективен.
Рекомендации:
Лечение ATTR-амилоидоза
До недавнего времени единственным методом лечения ATTR-амилоидоза была трансплантация печени, секретирующей нормальный транстиретин. Поскольку 98% всего сывороточного транстиретина синтезируется печенью, это позволяло прервать продукцию мутантного транстиретина. Трансплантация печени существенно замедляет прогрессирование ATTR-амилоидоза, а 20летняя выживаемость больных после трансплантации составляла 55,3% [36]. Однако уже имеющиеся массы амилоида способны выступать в роли ядра нуклеации для новых депозитов амилоида на основе нормального транстиретина (амилоидускоряющая субстанция). В настоящее время у больных с ранними стадиями ATTRамилоидной полиневропатии апробированы консервативные методы стабилизации тетрамерной структуры мутантного транстиретина и, следовательно, подавления его амилоидогенности. Один из таких препаратов – тафамидис замедлял на 52% (р=0,027) прогрессиро вание неврологических нарушений у больных ATTRамилоидозом, сохраняя функцию периферических соматических и автономных нервных волокон [12]. В клиническом исследовании III фазы лечение тафамидисом по сравнению с плацебо у больных с ATTR-амилоидозом сердца вызывало снижение общей смертности и частоты госпитализаций по сердечно-сосудистым причинам и задерживало ухудшение функциональной активности [81]. В Российской Федерации применение тафамидиса зарегистрировано только для лечения ATTR-амилоидоза с периферической полиневропатией.
В качестве стабилизатора транстиретина изучается также дифлюнизал из группы нестероидных противовоспалительных препаратов, однако его эффективность показана только в экспериментальных условиях.
Амилоидоз. Первичный (AL) и вторичный (AA). Виды амилоидоза.

Амилоидоз (амилоидная дистрофия) — гетерогенное заболевание, связанное с нарушением белкового обмена, сопровождающееся образованием и отложением в тканях специфического белково-полисахаридного комплекса — амилоида.
Амилоидоз может быть приобретенным или наследственным. Заболевание может быть локализованным или системным. Амилоид может аккумулироваться в печени, почках, сердце, нервах, и кровеносных сосудах, причиняя различные клинические синдромы, включая кардиомиопатию, гепатомегалию, протеинурию, макроглоссию, вегетативную дисфункцию, невропатию, почечную недостаточность, гипертензию.

Амилоидоз, Узел, Конго Красный
Выделяют множество видов амилоидоза, в частности:
В данной статье более подробно будут рассмотрены два наиболее часто встречающихся вида амилоидоза: первичный и вторичный.
Итак, что же такое первичный амилоидоз или AL-амилоидоз?
О пределение заболевания.
Амилоидный амилоидоз легкой цепи (AL), первичный системный амилоидоз (PSA) или просто первичный амилоидоз. Заболевание возникает, когда клетки человека, продуцирующие антитела (плазмоциты), не функционируют должным образом и производят аномальные белковые волокна из компонентов антител, называемых легкими цепями. Эти легкие цепи образуют амилоидные отложения, которые могут нанести серьезный ущерб различным органам. Аномальные легкие цепи в моче иногда называют «Белок Бенс-Джонса».
Эпидемиология.
AL-амилоидоз является наиболее распространенным типом системного амилоидоза в развитых странах с предполагаемой заболеваемостью 9 случаев на миллион жителей в год. Средний возраст диагностированных пациентов составляет 65 лет и менее 10% пациентов моложе 50 лет.
Признаки и симптомы.

Системный АL-амилоидоз. A.Макроглоссия с боковым гребешком языка. B. Двусторонняя периорбитальная пурпура. C.Псевдо-спортивный вид вторичных диффузных мышечных инфильтратов. D.Объемистая гепатомегалия из-за первичного печеночного амилоидоза. E.Диффузные двусторонние интерстициальные заболевания легких. F.Увеличение подчелюстной железы. Локализованный АL-амилоидоз. G. Узловой конъюнктивный амилоидоз. H.Горатнный амилоидный комок.
Диагностика заболевания.
Диагноз основывается на исследовании вовлеченного в патологический процесс участка, показывающего Конго красноположительные амилоидные отложения, которые окрашивают положительным антителом анти-LC иммуногистохимией и / или иммунофлуоресценцией. В связи с системным характером заболевания, неинвазивные биопсии, такие как аспирация брюшного жира, должны быть рассмотрены перед взятием биопсий из вовлеченных органов, чтобы уменьшить риск кровотечений.
Кроме того, можно исследовать кровь и мочу на наличие «легких цепей», которые могут формировать отложения амилоида, вызывая заболевание.
Лечение.
Наиболее эффективное лечение — аутологичные трансплантации костного мозга с помощью стволовых клеток. Однако не всем пациентам предписывают такой вид лечения.
Другие методы лечения могут включать применение химиотерапии, аналогичной той, которая используется при множественной миеломе. Комбинация мелфалана и дексаметазона была найдена эффективной у тех, кто не подходит для трансплантации стволовых клеток, и комбинация бортезомиба и дексаметазона в настоящее время широко распространена в клиническом использовании.
Прогноз.
Продолжительность жизни при АL-амилоидозе зависит от спектра поражения органов (основным фактором прогноза является амилоидная болезнь сердца), тяжести поражения отдельных органов и гематологического ответа на лечение.
Далее рассмотрим вторичный или AA-амилоидоз.
Определение заболевания.
Амилоидоз-АА также известен как вторичный амилоидоз, или реактивный внутрирастительный амилоидоз. Он возникает у пациентов с пролонгированными, хроническими воспалительными или инфекционными заболеваниями на протяжении нескольких лет.
У большинства пациентов с амилоидозом-АА обнаруживаются отложения амилоида в почках, что нарушает структуру почки и вызывает расстройство функции почки, наиболее часто проявляется появлением протеина в моче. В селезенке всегда имеются амилоидные отложения, количество которых может быть увеличено. Могут также быть отложения амилоида в других частях тела, которые обычно не вызывают симптомов.
Фибриллы обнаруженные в амилоидных отложениях образуются из белка, называемого амилоидным А белком, который является производным от нормального кровяного белка неизвестной функции, называемого белком сывороточного амилоида А (SАА). SАА, который вырабатывается в печени, содержится в очень малых количествах (менее 5 мг на литр) в крови всех здоровых людей. Но в ответ на множество видов воспаления, инфекции или ушибы тела, продукция SAA значительно увеличивается, вместе с продукцией другого нормального протеина — C-реактивного протеина (CRP). Производство некоторых других белков крови также увеличивается, хотя и в гораздо меньшей степени, как часть этой нормальной реакции на болезнь, которая называется реакцией острой фазы.
Какие заболевания могут повышать риск возникновения амилоидоза?
Воспалительные заболевания, которые чаще всего приводят к амилоидозу АА, относятся к следующим категориям.
Ревматологические заболевания, в том числе: ревматоидный артрит, ювенильный артрит, анкилозирующий спондилит и псориатический артрит.
Воспалительные заболевания желудочно-кишечного тракта, включая болезнь Крона и язвенный колит.
Хронические инфекции, такие как: туберкулез, бронхоэктатическая болезнь, остеомиелит, или инфекции, связанные с муковисцидозом, СПИД.
Злокачественные заболевания, включая болезнь Ходжкина, рак почки и болезни Кастельмана.
Наследственные расстройства, которые вызывают нарушение воспалительных генов, как: семейная Среднеземноморская лихорадка (FMF).
Как проявляется вторичный амилоидоз?
Наиболее распространенные симптомы амилоидоза АА вызваны отложениями амилоида в почках. Сначала отложения вызывают нарушение функции почек, а со временем могут привести к конечной стадии почечной недостаточности.
Симптомы болезни почек может включать в себя:
• протеинурия (белок в моче);
• потеря веса и ухудшение аппетита;
•повышенная потребность в мочеиспускании на ранних стадиях.
Амилоидоз АА обычно вызывает увеличение селезенки, которое часто не вызывает никаких симптомов, но может быть обнаружено при осмотре врачом. Редко: отложения амилоида АА в щитовидной железе могут вызвать зоб. Отложения амилоида АА в печени могут вызвать увеличение печени. Могут быть отложения амилоида АА в кишечнике и надпочечниках. Эти обычно не вызывает никаких симптомов, но иногда в редких случаях, они могут иметь серьезные последствия. Амилоид АА в сердце очень редко имеет клинически значимые эффекты.
Как диагностируют амилоидоз-АА?
Пациенты с амилоидозом АА обычно обращаются к врачу из-за нарушений функции почек. Более чем в 90% случаев, протеинурия (белок в моче) является первым признаком.
Таким образом, врачи могут заподозрить амилоидоз АА, если у пациента с давним воспалительным состоянием развиваются симптомы заболевания почек, особенно если есть также увеличенная селезенка. Другие признаки заболевания почек при амилоидозе АА могут включать:
Нефротический синдром:
— большое количество белка в моче (>3,5 г / сут);
— низкий уровень альбумина в крови;
— периферический отек – опухшие лодыжки.
Редко:
— гематурия (кровь в моче);
— дефекты почечных канальцев;
— нефрогенный несахарный диабет;
— кальцификация почек (отложения кальция в почках).
Диагноз амилоидоза может быть подтвержден (или может быть устранен) путем взятия биопсии из почки и / или прохождения сканирования SAP. Сканирование SAP показывает распределение и количество амилоида в органах по всему телу. Сканирование SAP произвело революцию в понимании естественного течения амилоидоза и его реакции на лечение.Биопсии могут показать микроскопические следы амилоида в небольших образцах тканей, но не могут обеспечить обзор всего тела. Сканирование SAP является единственным способом получить полную картину степени заболевания. При амилоидозе АА повторное последовательное сканирование SAP может быть очень полезно, показывая, что амилоидные отложения регрессируют (сокращаются), когда эффективно лечится основное воспалительное заболевание.
Лечение заболевания.
Лечение всех видов амилоидоза в настоящее время основано на следующих принципах:
1.Сокращение средств обеспечения для формирования амилоидного белка-предшественника(для этого применяют Колхицин).
2.Поддержание функций органов содержащих амилоид.
Подтип АА не лечится.