Что такое амавроз?
Амавроз — патология, которая сопровождается резким ухудшением зрения с быстрым его переходом в полную слепоту без видимых причин. Она не связана с болезнью глаз или их повреждением, а вызывается в результате снижения функции зрительного нерва. Есть несколько типов амавроза. Узнаем, каковы их причины и как они лечатся.

Амавроз на сегодняшний день изучен недостаточно. Врачи могут остановить развитие патологии или назначить лечение, но точные причины его возникновения пока неизвестны. Зачастую данный недуг является наследственным, но может быть и приобретенным (преходящим).
Преходящий (транзисторный) амавроз
При этой форме заболевания зрение пропадает временно. Причинами становятся следующие патологии:
При всех этих состояниях нарушается процесс передачи зрительных образов в мозг.
Врожденный амавроз

Его еще называют амаврозом Лебера. При этой патологии выявляются нарушения функций сетчатки. Они начинают развиваться еще в утробе. В сетчатой оболочке родопсин (зрительный пигмент) вырабатывается не полностью, а он отвечает за передачу информации от глаз к мозгу.
Симптомы
Основной симптом — резкая частичная или полная утрата зрения (обратимая и необратимая). Этот признак иногда сопровождается:

Также при амаврозе наблюдаются отеки глазных мышц, боли в глазах, возникают зрительные помехи. Симптомы будут зависеть от причин болезни. Вышеперечисленные признаки сложно определить, когда диагноз нужно поставить ребенку до 3-4 лет, так как он не может описать свое состояние. Врач обращает внимание на такие симптомы, как:
Эти признаки способны заметить и родители. Они должны в этом случае быстрее обратиться к врачу, чтобы избежать осложнений недуга.
Лечение
Лечения амавроза Лебера сегодня не существует. Транзисторную форму можно вылечить путем установления причин болезни и их устранения. Важно вовремя заметить ухудшение зрения и обратиться к специалисту. Самое серьезное осложнение амавроза — полная и необратимая слепота.
Амавроз Лебера

Амавроз Лебера – это наследственное заболевание, характеризующееся врожденным поражением светочувствительных клеток сетчатки глаза и в некоторых случаях другими общими нарушениями (аномалии почек, ЦНС). При этой патологии в первые месяцы жизни ребенка или сразу после рождения появляется нистагм, ослабление или отсутствие реакции зрачка на свет. В дальнейшем ребенок может тереть глаза (симптом Франческетти), возникает дальнозоркость и светобоязнь, возможна полная потеря зрения. Диагностика основывается на данных осмотра пациента врачом-офтальмологом, электроретинографии, исследования наследственного анамнеза и генетических анализов. Специфическое лечение амавроза Лебера на сегодняшний день не разработано.
МКБ-10
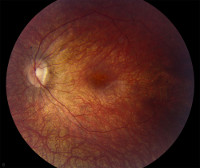
Общие сведения
Врожденный амавроз Лебера представляет собой гетерогенную группу заболеваний, причиной которых выступают мутации в 18 генах, кодирующих различные белки сетчатки, в том числе опсин. Впервые амавроз был описан еще в XIX веке (в 1867 году) Т. Лебером, указавшим основные проявления этого заболевания – маятниковый нистагм, слепота, появление пигментных пятен и включений на глазном дне. Средняя распространенность заболевания составляет 3:100000 населения.
Амавроз Лебера в равной мере поражает как мужчин, так и женщин. Заболевание составляет примерно 5% от всех наследственных ретинопатий. Современная генетика разрабатывает методики лечения данной патологии, имеются обнадеживающие результаты генной терапии одной из форм амавроза Лебера, обусловленной мутацией в гене RPE65. Отдельно выделяют атрофию зрительных нервов Лебера, которая также характеризуется постепенной потерей остроты зрения и впоследствии полной слепотой. Однако это заболевание совершенно другой генетической природы и обусловлено повреждением митохондриальной ДНК, которая имеет свой уникальный тип наследования (по материнской линии).
Причины
Основной механизм расстройства зрения при амаврозе Лебера – нарушение метаболизма в палочках и колбочках, которое ведет к летальным повреждениям фоторецепторов и их разрушению. Однако непосредственная причина таких изменений различается в зависимости от того, мутация какого именно гена вызвала заболевание.
Один из наиболее распространенных типов амавроза Лебера (тип 2, LCA2) обусловлен наличием мутантного гена RPE65 на первой хромосоме. Известно более 80-ти мутаций этого гена, некоторые из которых, помимо амавроза Лебера, вызывают и определенные формы пигментной абиотрофии сетчатки. Белок, кодируемый PRE65, отвечает за метаболизм ретинола в пигментном эпителии сетчатой оболочки глаза, поэтому при наличии генетического дефекта этот процесс нарушается с развитием побочных метаболических путей. В результате этого синтез родопсина в фоторецепторах прекращается, что и приводит к характерной клинической картине заболевания. Мутантные формы гена наследуются по аутосомно-рецессивному механизму.
Менее распространенная форма амавроза Лебера (тип 14) вызвана мутацией гена LRAT на 4-й хромосоме. Он кодирует белок лецитин-ретинол-ацилтрансферазу, который располагается в микросомах гепатоцитов и обнаружен в сетчатке глаза. Этот фермент участвует в метаболизме ретиноидов и витамина А, из-за наличия мутаций в гене полученный протеин не может полноценно выполнять свои функции, из-за чего развивается дегенерация фоторецепторов, которая клинически проявляется амаврозом Лебера или ювенильной пигментной абиотрофией сетчатки. Имеет аутосомно-рецессивный характер наследования.
Амавроз Лебера тип 8 наиболее часто приводит к врожденной слепоте, ответственный за развитие этой формы заболевания ген CRB1 располагается на 1-й хромосоме и имеет аутосомно-рецессивный характер наследования. При этом выяснено, что кодируемый данным геном белок принимает непосредственное участие в эмбриональном развитии фоторецепторов и пигментного эпителия сетчатки. Более точных данных по патогенезу данной формы амавроза Лебера на сегодняшний день не накоплено. Аналогичная ситуация с мутацией гена LCA5, расположенного в 6-й хромосоме и ассоциированного с 5-м типом амавроза. В настоящее время выявлен только белок, кодируемым данным геном – леберцилин, но его функции в сетчатке непонятны.
Также выявлено две формы амавроза Лебера, которые наследуются по аутосомно-доминантному механизму – тип 7, обусловленный мутацией гена CRX, и тип 11, ассоциированный с нарушением гена IMPDH1. Ген CRX кодирует белок, который обладает множеством функций – контроль развития фоторецепторов в эмбриональный период, поддержание их адекватного уровня во взрослом возрасте, участие в синтезе других протеинов сетчатки (является фактором транскрипции). Поэтому в зависимости от характера мутации гена CRX клиника амавроза Лебера 7-го типа может быть разнообразной – от врожденной слепоты до относительно позднего и вялотекущего ухудшения зрения.
Инозин-5′-монофосфатдегидрогеназа 1, кодируемый геном IMPDH1, представляет собой фермент, регулирующий рост клеток и образование нуклеиновых кислот, однако это пока не позволяет прояснить патогенез того, как нарушения этого белка приводят к 11-му типу амавроза Лебера.
Классификация амавроза Лебера
В настоящее время полностью доказана взаимосвязь между клиническими проявлениями и мутациями определенных генов для 16-ти типов амавроза Лебера. Также имеются указания об открытии еще двух генов, повреждения в которых приводят к такому заболеванию, но пока в этом отношении проводятся дополнительные исследования.
Кроме того, иногда в клинической классификации выделяют не только название поврежденного гена, но и характер мутации, поскольку это имеет значительное влияние на течение амавроза Лебера. Более того, различные типы мутаций в одном и том же гене могут приводить к совершенно разным заболеваниям – например, некоторые разновидности делеций в гене CRX могут приводить не к амаврозу, а к палочко-колбочковой дистрофии. Некоторые мутации генов RPE65, LRAT и CRB1 являются причиной различных форм пигментной абиотрофии сетчатки.
Симптомы амавроза Лебера
Симптоматика амавроза Лебера достаточно вариабельна и зависит от типа заболевания и характера мутации гена. В большинстве случаев при рождении ребенка патология не определяется – даже при осмотре глазного дна изменения наблюдаются лишь в нескольких процентах случаев. По мере его роста родители могут замечать, что ребенок не задерживает взгляд на предметах и окружающих, а в более старшем возрасте может болезненно реагировать на свет (появляется фотофобия), часто тереть глаза и указывать на них пальцем (симптом Франческетти, окулопальцевый синдром). Обнаруживается нистагм, который возникает еще в первые 2-3 месяца жизни и часто является одним из первых проявлений амавроза Лебера, замедленная реакция зрачка на свет или ее полное отсутствие.
В ряде случаев наблюдается врожденная слепота. Если же ребенок родился с относительно сохранной функцией зрения, то в первые годы жизни, помимо указанных симптомов, у его также развивается дальнозоркость, косоглазие, сильно страдает острота зрения. Обычно к 10-ти годам большинство больных с амаврозом Лебера полностью слепнут. В дальнейшем у них могут возникать и другие нарушения зрительного аппарата – кератоконус, катаракта, глаукома. При некоторых типах заболевания могут наблюдаться и сопутствующие нарушения – поражения ЦНС, глухота.
Диагностика
В современной офтальмологии диагностика амавроза Лебера производится на основании осмотра глазного дна, мониторинга динамики изменений в нем, данных электроретинографии. Немаловажную роль играет также изучение наследственного анамнеза, а для некоторых типов заболевания – генетическое секвенирование последовательности ключевых генов.
Дифференциальную диагностику производят с различными формами пигментной абиотрофии сетчатки (при ней сохраняется нормальная или немного сниженная амплитуда волн на электроретинограмме) и некоторыми типами атрофии зрительных нервов.
Лечение амавроза Лебера
На сегодняшний день специфического лечения любого типа амавроза Лебера не существует. На этапе клинических испытаний находится генно-инженерное введение гена RPE65 в сетчатую оболочку глаза больных амаврозом 2-го типа, имеются первые данные о значительном улучшении зрения подопытных больных. В случае же остальных форм заболевания такого прогресса пока нет. Поддерживающее лечение сводится к витаминной терапии, внутриглазным инъекциям сосудорасширяющих средств. При дальнозоркости назначается ношение очков.
Прогноз
В плане сохранения зрения прогноз крайне неблагоприятный, практически 95% больных полностью теряют способность видеть к 10-му году жизни. Кроме того, это наследственное заболевание может осложняться проблемами с ЦНС, почками, эндокринной системой, что требует более тщательного медицинского мониторинга для своевременного выявления подобных нарушений.
Что такое амавроз
 Амавроз – медицинский термин, означающий потерю или ухудшение зрения, при котором видимое повреждение структур глаза отсутствует.
Амавроз – медицинский термин, означающий потерю или ухудшение зрения, при котором видимое повреждение структур глаза отсутствует.
Чаще всего эпизоды временной слепоты случаются из-за нарушения кровообращения в области головы и шеи.
Зрение при этом восстанавливается. Прогрессирующее ухудшение зрения генетической природы, так называемый амавроз Лебера, приводит к полной слепоте.
Причины
Внезапная кратковременная потеря зрения или его ухудшение могут быть вызваны нарушением кровоснабжения головного мозга, неврологическими заболеваниями.
Среди наиболее частых этиологических факторов рассматривают:
Причинами преходящей потери зрения могут быть некоторые болезни глаза и его орбиты:
В некотором количестве случаев причина остается неустановленной.
Если речь идет о генетически обусловленном заболевании, вызывающем постепенное снижение зрение вплоть до полной его утраты, то причина кроется в прогрессирующей гибели палочек и колбочек сетчатки глаза.
Группа риска
Люди, страдающие сердечно-сосудистыми заболеваниями, диабетики, больные остеохондрозом могут испытывать кратковременные приступы потери зрения.
Пожилой возраст, курение и неправильное питание, провоцирующее развитие атеросклероза – вот некоторые факторы, увеличивающие риск возникновения подобного состояния.
В группе риска находится потомство тех людей, у кого в семейном анамнезе были случаи прогрессирующей с раннего возраста потери зрения, приводящее к слепоте. Генетический фактор необходимо учитывать при ответственном планировании семьи.
Формы амавроза
Описывают две основные формы заболевания: врожденный амавроз Лебера и преходящий амавроз фугас.
 Врожденный амавроз Лебера – наследственное заболевание, вызванное мутациями генов, отвечающих за способность светочувствительных клеток сетчатки глаза к восстановлению. Приводит к атрофии сетчатой оболочки глаза, постепенному ухудшению зрения и полной слепоте. Описано в XIX в. немецким офтальмологом Теодором Лебером.
Врожденный амавроз Лебера – наследственное заболевание, вызванное мутациями генов, отвечающих за способность светочувствительных клеток сетчатки глаза к восстановлению. Приводит к атрофии сетчатой оболочки глаза, постепенному ухудшению зрения и полной слепоте. Описано в XIX в. немецким офтальмологом Теодором Лебером.
Существуют данные о том, что хинин, принимаемый в больших дозах, в виду своего токсического действия, вызывает амавроз.
Врожденный
Амавроз Лебера — наследственная болезнь, проявляется с рождения или в первые месяцы жизни. К 25-30 годам у больного почти неизбежно развивается слепота. Довольно редкое заболевание: поражает одного из 40 тыс. новорожденных. Нарушение вызвано неправильным развитием фоторецепторов (светочувствительных сенсорных нейронов сетчатки глаза: колбочек и палочек) и недостаточностью родопсина, отвечающего за преобразование света в нервные импульсы.
Куриная слепота наблюдается с самого детства. Светочувствительные клетки постоянно отмирают, при этом, вследствие генных мутаций, потеряна их способность к восстановлению. Сначала человек способен ориентироваться при ярком освещении, но в полумраке он уже беспомощен. Сильно сужается поле зрения: остается участок центрального зрения вокруг центра. Постепенно зрение пропадает полностью.
Насчитывается от 11-и до 18-и форм заболевания. Такое многообразие форм объясняется тем, что мутации генов, отвечающих за регенерацию светочувствительного пигмента, разнообразны.
Симптомы общие для всех видов наследственного заболевания: нистагм, слабый зрачковый рефлекс или его отсутствие, значительная потеря зрения или полная слепота.
Имеются данные об успешном лечении одной из форм болезни, вызванной мутацией гена RPE65, с помощью генной инженерии.
Преходящий
Симптомы амавроза
 Амавроз Лебера обычно диагностируется в раннем детстве и проявляется следующими признаками:
Амавроз Лебера обычно диагностируется в раннем детстве и проявляется следующими признаками:
Диагностика амавроза
Все, кто испытывал резкое падение зрения или его утрату, обязательно должны быть осмотрены специалистом. Необходимо выяснить причину данного состояния, так как любая форма потери зрения указывает на серьезные системные нарушения или заболевания глаз. К примеру, эпизод амавроза может быть симптомом надвигающегося инсульта или инфаркта.
Лечение амавроза
Врожденный амавроз в большинстве случаев — неизлечимое заболевание, несмотря на некоторые успехи генной инженерии.
Лечение случаев временной слепоты зависит от причины, ее вызвавшей — от основного заболевания. Например, лечение пациентов, испытавших внезапную потерю зрения вследствие сосудистых нарушений, заключается, в том числе, в назначении лекарств, влияющих на вязкость крови. Такие лекарства, как аспирин уменьшают способность тромбоцитов к склеиванию. Целесообразно назначение средств, понижающих давление. В некоторых случаях показано удаление поврежденного или закупоренного участка сонной артерии.
Осложнения амавроза
Эпизоды преходящей слепоты приводят к нарушениям в ретинальной ткани, наблюдаются сужение и спазм сосудов, мелкие сосудистые кровоизлияния.
Прогноз
Врожденный амавроз приводит к полной слепоте. Уже в молодом возрасте человек становится инвалидом.
При успешном лечении основного заболевания, вызвавшего эпизод временной утраты зрения, прогноз благоприятен.
Профилактика
При целом ряде неврологических и сердечно-сосудистых заболеваний, атеросклерозе, сахарном диабете чтобы избежать негативного влияния на зрение, необходимо соблюдать установленный режим лечения. Отказ от вредных привычек — курение и алкоголь — правильное питание также способствуют сохранению хорошего состояния глаз.
Проведение генетического анализа перед вступлением в брак поможет уменьшить вероятность рождения больного потомства.
Полезное видео
Публикации в СМИ
Заболевания сетчатки и зрительного нерва наследственные
Наследственные заболевания сетчатки и зрительного нерва — обширная гетерогенная группа заболеваний, приводящих к снижению или полному отсутствию зрения. В зависимости от преимущественного поражения сетчатки или зрительного нерва выделяют следующие подгруппы.
• Амавроз врождённый — наследуемая абиотрофия палочек и колбочек сетчатки и атрофия зрительных нервов, проявляющиеся быстрым развитием двусторонней центральной скотомы. Наблюдают преимущественно у мужчин, может развиться в любом возрасте (многие формы врождённые — амавроз врождённый Лебера: • тип 1: 204000, GUC2D, GUCSD, LCA1, 600179 [гуанилат циклаза 2D], 17p13; • тип 2: 204100, RPE65 [белок пигментного эпителия сетчатки], 180069, 1p31). Характеризуется прогрессирующим ухудшением зрения. В основе лежат наследуемые заболевания (мутации митохондриального и ядерного геномов). Известно, как минимум, 18 мутаций митохондриальной ДНК и не менее 6 локусов ядерной ДНК. Большую частоту заболевания у мужчин объясняют влиянием андрогенов (например, описан случай резкой потери зрения больным с мутацией MTND4*LHON11778А после терапии андрогенами).
• Атрофия зрительного нерва — конечный результат дистрофических изменений ганглиозных нейронов сетчатки. Первичная атрофия зрительного нерва — генетически обусловленная патология; компонент или единственное проявление при ряде наследственных заболеваний, отличающихся по срокам появления атрофии, степени тяжести, сопутствующим симптомам и типу наследования. Отмечают все основные типы наследования, включая мутации митохондриального генома (атрофия зрительного нерва Лебера, #535000, гены MTND [NADH-убихинон оксидоредуктаза митохондриального генома, КФ 1.6.5.3]).
• Атрофия зрительного нерва — часто компонент наследственных синдромов •• Атаксия Фридрайха с атрофией зрительного нерва и нейросенсорной тугоухостью (136600, Â ) •• Болезнь ван Бухема: иногда первое проявление этой патологии — атрофия зрительного нерва за счёт его сдавления гиперостозными разрастаниями костей черепа •• Атрофия зрительного нерва, глухота и дистальная нейрогенная амиотрофия (258650, r ) •• Метафизарная дисплазия, анетодермия (фокальная атрофия кожи) и атрофия зрительного нерва (250450, r ) •• Атрофия зрительного и слухового нервов с деменцией (311150, À ).
• Дегенерация сетчатки пигментная — прогрессирующая атрофия нейроэпителия с атрофией и пигментной инфильтрацией внутренних слоёв сетчатки. Часто применяемый термин «пигментный ретинит» не совсем точно отражает суть процесса, поскольку воспалительной реакции как таковой не возникает. Основные проявления обусловлены утратой палочек сетчатки. В основе лежит множество мутаций в ряде локусов (например, гены периферина [179605], родопсина [180380], цГМФ управляемого канала фоторецепторов [123825] и др.). Пигментная дегенерация сетчатки, не ассоциированная с другими нарушениями, наиболее часто наследуется как r (268000) и реже как À рецессивная (312600) признак. Доминантное наследование отмечают в 3–4% случаев. Кроме того, атипичный пигментный ретинит наблюдают при множестве других состояний, включая такие рецессивные нарушения, как абеталипопротеинемия (200100), синдромы Альстрема (203800), Рефсума (266500), Барде–Бидла (209900), Лоуренса–Муна (245800), Ашера (276900), Коккейна (216400), паллидарная дегенерация (260200).
• Дегенерация сетчатки коллоидная (сотовидный хориоидит Дойна, *126600, 2p16, ген DHRD, Â ). Множественные очаги округлой формы беловатого цвета, расположенные у диска зрительного нерва и в зоне между верхней и нижней височными артериолами.
• Дистрофия глазного дна Сорсби (#136900, псевдовоспалительная дистрофия глазного дна, 22q12.1–q13.2, ген тканевого ингибитора металлопротеиназы-3 TIMP3 [*188826], Â ). Клинически: дистрофия жёлтого пятна, глазного дна, атрофия сетчатки.
• Дистрофия жёлтого пятна кистозная (*153880, 7p21–p15, ген MDDC, Â ). Клинически: кистозный отёк жёлтого пятна, беловатые точечные отложения в стекловидном теле, дальнозоркость, косоглазие, снижение остроты зрения, перицентральный пигментный ретинит.
• Дистрофия палочек и колбочек, типы и гены: • тип 1, 600624, CORD1, CRD1, 18q21.1 q21.3; • тип 2, 120970, CRX, CORD2, CRD, 602225, 19q13.3
• Дистрофия пигментного эпителия сетчатки, 179605 (мутации гена периферина RDS, RP7), 6p21.1 cen
• Дистрофия сетчатки ранняя (аутосомно рецессивная): 180069, ген RPE65, 1p31
• Дистрофия хориоидеи центральная (215500, ген CACD, 17p)
Дифференциальная диагностика • Инфекционные ретинопатии — вирусные (краснуха) или бактериальные (сифилис) • Остаточные явления экссудативной отслойки сетчатки • Вторичные токсические ретинопатии (хлорохин или фенотиазины) • Другие патологические состояния (окклюзия глазной артерии, травмы).
Течение и прогноз • Прогноз наиболее неблагоприятный при врождённых формах • Течение в большинстве случаев хроническое, медленно прогрессирующее.
МКБ-10 • H31.2 Наследственная дистрофия сосудистой оболочки глаза • H35.5 Наследственные ретинальные дистрофии • Q14 Врождённые аномалии [пороки развития] заднего сегмента глаза
ПРИЛОЖЕНИЯ
Слепота цветовая (ахроматопсия) — отсутствие цветового зрения. Возможность различать любые цвета (трихромазия, основы теории цветного зрения предложил в 1802 г. Томас Янг) определяется присутствием в сетчатке всех трёх зрительных пигментов (для красного, зелёного и синего — первичные цвета). Дихромазии — дефекты цветового восприятия (преимущественно у мужчин; например, разные дефекты у мужчин составляют 8% общей популяции) по одному из первичных цветов — подразделяют на протанопии, дейтанопии и тританопии (от греч. первый, второй и третий [имеются в виду порядковые номера первичных цветов: соответственно красный, зелёный, синий]) • Протанопия (страдает восприятие красного, примерно 25% случаев цветовой слепоты) • Дейтанопия (цветовая слепота по восприятию зелёного, около 75% всех случаев) • Тританопия (страдает преимущественно восприятие фиолетового цвета, дефектное зрение по синему и жёлтому) • Дальтонизм — нарушение цветового зрения — неспособность различать красный и зеленый цвета. МКБ-10. H53.5 Аномалии цветового зрения.
Слепота преходящая (amaurosis fugax) — острый эпизод полной или частичной потери зрения, длящийся обычно не более 10 мин. Причины: кратковременная ишемия в бассейне сонной артерии (как правило, эмболия артерий сетчатки). Лечение • Лекарственная терапия •• Антигипертензивные средства •• Фибринолитические средства •• Антиагреганты (ацетилсалициловая кислота, дипиридамол) • Хирургическое: эндартерэктомия сонной артерии. МКБ-10. G45.3 Преходящая слепота.
Хориоидеремия Х-сцепленная (*303100, ген CHM, Xq21.2; форма с глухотой и ожирением, 303110) — наследственная болезнь глаз, прогрессирующие понижение остроты зрения, сужение полей зрения, гемералопия и близорукость; характерные изменения глазного дна (почти полное отсутствие сосудистого рисунка, резкие контуры и красновато-коричневый цвет жёлтого пятна, нечёткость границ диска зрительного нерва). Синоним: атрофия сосудистой оболочки прогрессирующая. МКБ-10. H31.2 Наследственные дистрофии сосудистой оболочки глаза.
Код вставки на сайт
Заболевания сетчатки и зрительного нерва наследственные
Наследственные заболевания сетчатки и зрительного нерва — обширная гетерогенная группа заболеваний, приводящих к снижению или полному отсутствию зрения. В зависимости от преимущественного поражения сетчатки или зрительного нерва выделяют следующие подгруппы.
• Амавроз врождённый — наследуемая абиотрофия палочек и колбочек сетчатки и атрофия зрительных нервов, проявляющиеся быстрым развитием двусторонней центральной скотомы. Наблюдают преимущественно у мужчин, может развиться в любом возрасте (многие формы врождённые — амавроз врождённый Лебера: • тип 1: 204000, GUC2D, GUCSD, LCA1, 600179 [гуанилат циклаза 2D], 17p13; • тип 2: 204100, RPE65 [белок пигментного эпителия сетчатки], 180069, 1p31). Характеризуется прогрессирующим ухудшением зрения. В основе лежат наследуемые заболевания (мутации митохондриального и ядерного геномов). Известно, как минимум, 18 мутаций митохондриальной ДНК и не менее 6 локусов ядерной ДНК. Большую частоту заболевания у мужчин объясняют влиянием андрогенов (например, описан случай резкой потери зрения больным с мутацией MTND4*LHON11778А после терапии андрогенами).
• Атрофия зрительного нерва — конечный результат дистрофических изменений ганглиозных нейронов сетчатки. Первичная атрофия зрительного нерва — генетически обусловленная патология; компонент или единственное проявление при ряде наследственных заболеваний, отличающихся по срокам появления атрофии, степени тяжести, сопутствующим симптомам и типу наследования. Отмечают все основные типы наследования, включая мутации митохондриального генома (атрофия зрительного нерва Лебера, #535000, гены MTND [NADH-убихинон оксидоредуктаза митохондриального генома, КФ 1.6.5.3]).
• Атрофия зрительного нерва — часто компонент наследственных синдромов •• Атаксия Фридрайха с атрофией зрительного нерва и нейросенсорной тугоухостью (136600, Â ) •• Болезнь ван Бухема: иногда первое проявление этой патологии — атрофия зрительного нерва за счёт его сдавления гиперостозными разрастаниями костей черепа •• Атрофия зрительного нерва, глухота и дистальная нейрогенная амиотрофия (258650, r ) •• Метафизарная дисплазия, анетодермия (фокальная атрофия кожи) и атрофия зрительного нерва (250450, r ) •• Атрофия зрительного и слухового нервов с деменцией (311150, À ).
• Дегенерация сетчатки пигментная — прогрессирующая атрофия нейроэпителия с атрофией и пигментной инфильтрацией внутренних слоёв сетчатки. Часто применяемый термин «пигментный ретинит» не совсем точно отражает суть процесса, поскольку воспалительной реакции как таковой не возникает. Основные проявления обусловлены утратой палочек сетчатки. В основе лежит множество мутаций в ряде локусов (например, гены периферина [179605], родопсина [180380], цГМФ управляемого канала фоторецепторов [123825] и др.). Пигментная дегенерация сетчатки, не ассоциированная с другими нарушениями, наиболее часто наследуется как r (268000) и реже как À рецессивная (312600) признак. Доминантное наследование отмечают в 3–4% случаев. Кроме того, атипичный пигментный ретинит наблюдают при множестве других состояний, включая такие рецессивные нарушения, как абеталипопротеинемия (200100), синдромы Альстрема (203800), Рефсума (266500), Барде–Бидла (209900), Лоуренса–Муна (245800), Ашера (276900), Коккейна (216400), паллидарная дегенерация (260200).
• Дегенерация сетчатки коллоидная (сотовидный хориоидит Дойна, *126600, 2p16, ген DHRD, Â ). Множественные очаги округлой формы беловатого цвета, расположенные у диска зрительного нерва и в зоне между верхней и нижней височными артериолами.
• Дистрофия глазного дна Сорсби (#136900, псевдовоспалительная дистрофия глазного дна, 22q12.1–q13.2, ген тканевого ингибитора металлопротеиназы-3 TIMP3 [*188826], Â ). Клинически: дистрофия жёлтого пятна, глазного дна, атрофия сетчатки.
• Дистрофия жёлтого пятна кистозная (*153880, 7p21–p15, ген MDDC, Â ). Клинически: кистозный отёк жёлтого пятна, беловатые точечные отложения в стекловидном теле, дальнозоркость, косоглазие, снижение остроты зрения, перицентральный пигментный ретинит.
• Дистрофия палочек и колбочек, типы и гены: • тип 1, 600624, CORD1, CRD1, 18q21.1 q21.3; • тип 2, 120970, CRX, CORD2, CRD, 602225, 19q13.3
• Дистрофия пигментного эпителия сетчатки, 179605 (мутации гена периферина RDS, RP7), 6p21.1 cen
• Дистрофия сетчатки ранняя (аутосомно рецессивная): 180069, ген RPE65, 1p31
• Дистрофия хориоидеи центральная (215500, ген CACD, 17p)
Дифференциальная диагностика • Инфекционные ретинопатии — вирусные (краснуха) или бактериальные (сифилис) • Остаточные явления экссудативной отслойки сетчатки • Вторичные токсические ретинопатии (хлорохин или фенотиазины) • Другие патологические состояния (окклюзия глазной артерии, травмы).
Течение и прогноз • Прогноз наиболее неблагоприятный при врождённых формах • Течение в большинстве случаев хроническое, медленно прогрессирующее.
МКБ-10 • H31.2 Наследственная дистрофия сосудистой оболочки глаза • H35.5 Наследственные ретинальные дистрофии • Q14 Врождённые аномалии [пороки развития] заднего сегмента глаза
ПРИЛОЖЕНИЯ
Слепота цветовая (ахроматопсия) — отсутствие цветового зрения. Возможность различать любые цвета (трихромазия, основы теории цветного зрения предложил в 1802 г. Томас Янг) определяется присутствием в сетчатке всех трёх зрительных пигментов (для красного, зелёного и синего — первичные цвета). Дихромазии — дефекты цветового восприятия (преимущественно у мужчин; например, разные дефекты у мужчин составляют 8% общей популяции) по одному из первичных цветов — подразделяют на протанопии, дейтанопии и тританопии (от греч. первый, второй и третий [имеются в виду порядковые номера первичных цветов: соответственно красный, зелёный, синий]) • Протанопия (страдает восприятие красного, примерно 25% случаев цветовой слепоты) • Дейтанопия (цветовая слепота по восприятию зелёного, около 75% всех случаев) • Тританопия (страдает преимущественно восприятие фиолетового цвета, дефектное зрение по синему и жёлтому) • Дальтонизм — нарушение цветового зрения — неспособность различать красный и зеленый цвета. МКБ-10. H53.5 Аномалии цветового зрения.
Слепота преходящая (amaurosis fugax) — острый эпизод полной или частичной потери зрения, длящийся обычно не более 10 мин. Причины: кратковременная ишемия в бассейне сонной артерии (как правило, эмболия артерий сетчатки). Лечение • Лекарственная терапия •• Антигипертензивные средства •• Фибринолитические средства •• Антиагреганты (ацетилсалициловая кислота, дипиридамол) • Хирургическое: эндартерэктомия сонной артерии. МКБ-10. G45.3 Преходящая слепота.
Хориоидеремия Х-сцепленная (*303100, ген CHM, Xq21.2; форма с глухотой и ожирением, 303110) — наследственная болезнь глаз, прогрессирующие понижение остроты зрения, сужение полей зрения, гемералопия и близорукость; характерные изменения глазного дна (почти полное отсутствие сосудистого рисунка, резкие контуры и красновато-коричневый цвет жёлтого пятна, нечёткость границ диска зрительного нерва). Синоним: атрофия сосудистой оболочки прогрессирующая. МКБ-10. H31.2 Наследственные дистрофии сосудистой оболочки глаза.